818
Ключ до розуміння інсулінорезистентності
Тема інсулінорезистентності дуже гостра. Більшість захворювань цивілізації, починаючи від депресії, серцево-судинних захворювань до хвороби Альцгеймера, асоціюються з метаболічними розладами. Однак дуже цікаво, що з'єднання існує між життям і захворюваннями, як поживні речовини викликають специфічні порушення метаболізму.
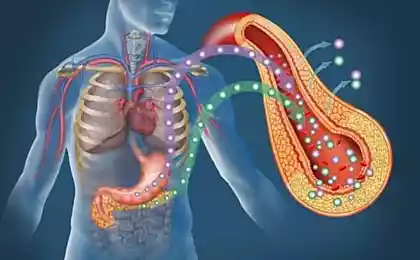
Тому сьогодні я буду говорити про FFA - безкоштовні жирні кислоти (у ілюстраціях, позначених FFA - безкоштовні жирні кислоти). З одного боку, FFA є основним джерелом енергії, але з іншого боку, це важлива молекула сигналізації.
Я особливо хочу відзначити, що коли мова йде про хронічне збільшення ФФА як причину інсулінорезистентності (ІР), то пам'ятайте, що проблема не ФФА, але що вони ніде використовувати, тому їх надлишки з'являються в крові. Ця стаття є довгою, але хто цікавиться розумінням логіки здоров'я та хвороби.
Безкоштовні жирні кислоти: Ключ до розуміння
Безкоштовні жирні кислоти.
В результаті гідролізу тригліцеридів, що містяться в жирових тканинах. Плазма жирні кислоти є естерифікованими і в цьому випадку є переважно альбумінними або не естерифікованими і безкоштовні. Безкоштовні жирні кислоти (FFA) є частиною 5 ліпопротеїнів.
Останній складається з довгоочіданих жирних кислот, міцно зв'язаних з двома певними регіонами молекули альбуміну; при збільшенні рівня ФФА в плазмі вони займають додаткові ділянки цієї молекули, але зв'язування в цьому випадку менш сильний.
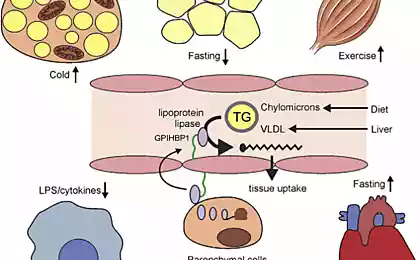
FFAs є основною енергетичною субстратністю тіла. Вони утворюються в процесі ліполізу тригліцеридів, накопичених в клітинах жирової тканини. Ліза тканин в цих клітинах під нейроендокринним контролем і активується через систему циклази аденілат.
Друге джерело плазми FFA є гідролізом тригліцеридів, що містяться в ліпопротеїнів, під впливом ліпопротеїну ліпоза. Уміння скелетних м’язів (та інших тканин) для регулювання їх метаболізму до домінантою субстрату в даний час зазвичай називають «добрим метаболічним здоров’ям» або «метаболічною гнучкістю». Зрозуміло, що хороше «метаболічне здоров’я» пов’язане з нормальною чутливістю до інсуліну.
Безкоштовний обмін жирною кислотою.
Період напіврозпаду ФФА дуже короткий - 4 - 8 хвилин, і вони легко поглинаються з плазми м'язовими клітинами тіла. Другим способом їх обміну є всмоктування печінки і ресинтезу в тригліцериди, які потім можна перевозити з печінки в складі VLDL або окислюється до ацетил-CoA. У фізіологічних умовах рівень ФФА в крові може швидко зростатися і знизитися, наповнювати потреби організму для цієї форми енергії.
Вміст їх зазвичай нижчий після поглинання вуглеводів і отриманого випуску інсуліну, але в міру зниження рівня глюкози в крові після їжі їх рівень збільшується. На порожньому шлунку крові міститься, як правило, 400 - 600 мкЕк / л ФФА; з більшою швидкістю (до 24 - 72 годин), рівень ФФА може досягати 1000 - 1500 мкЕк / л. Глюкагон, адреналін, гормону росту і АКТ також підвищують рівень ФФА. Основними фізіологічними регуляторами вмісту ФФА в плазмі є інсулін і адреналін.
Кожна хвилина, 20-40% ФФА, що надходить до плазми, вводяться окислені, рестеризовані або перетворюються на інші жирні кислоти. Під час відпочинку окислення відбувається переважно в печінці і серці, а під час стресу - в скелетних м'язах, а в останньому випадку частка окислюється ФФА збільшується від близько 20 до 60%.
Більшість ФФА, захоплених клітинами печінки, переоцінюються для формування переважно тригліцеридів, а також фосфоліпідів, для синтезу яких зазвичай використовується лінолеєва кислота. У плазмі FFAs міститься в діапазоні концентрацій від 100 мкмоль / л до 1 ммоль / л і їх рівень сильно залежить від часу доби.
Після кожної їжі рівень ФФА в плазмі осені, так як інсулін пригнічує ліполіз у жирових клітинах, в результаті чого ФФА утворюється. На ніч збільшується концентрація ФФА в плазмі. Для цих нормальних щоденних коливань в рівні ФФА практично всіх інших тканин, зокрема, скелетних м'язів, «свербіж» від використання глюкози (день) до споживання ФФА (нині).
Деякі причини порушення обміну ФФА.
Ключовим порушенням метаболізму вільних жирних кислот є їх хронічне збільшення. Хронічне висіння рівнів ФФА може бути викликане різними причинами харчової та стрес-типу. Наприклад, надлишок вуглеводів, в яких починається синтез нових жирних кислот від зайвих вуглеводів.
З 75-80% глюкози у здоровій людини вкладається скелетний м'яз, фізична активність робітників може запобігти розвитку «функціональної» інсулінорезистентності. Під час фізичного навантаження також викликає збільшення ФФА.
Хронічний стрес будь-якого типу є ще однією причиною хронічного збільшення ФФА. Під час стресу в крові рівень ФФА підвищується, щоб забезпечити роботу серця і м'язів. Але людина зазвичай не рухається і всі ці жирні кислоти циркулюють в крові протягом тривалого часу.
Великомасштабними дослідженнями стали неперервні зв’язки між становищем людини на соціально-економічній сході та ймовірність розвитку метаболічного синдрому. Авторами висновку, що розвиток метаболічного синдрому є біологічним механізмом, що призводить до «соціальної нерівності в розподілі коронарного ризику серед людей. й
Високий коронарний ризик (завищені рівні тригліцериду і низькі рівні холестерину ліпопротеїну високої щільності) пов'язані з низьким соціально-економічним статусом (дорослим), що в дорослому віці призводить до зайвої ваги і ожиріння.
Крім того, психологічний стрес також сприяє розвитку T1D і T2D, так як високі рівні ФФА асоціюються з підвищеним утворенням в мітохондрії реактивних видів кисню (окислювальний стрес), що в кінцевому рахунку призводить до зменшення здатності клітин реагувати на адекватно дії інсуліну.
FFA - це метаболіт і молекула сигналізації.
Чисельні дослідження в останні роки показали, що, по-перше, FFA не тільки високоенергетичне паливо, але й важливі молекули сигналізації. Їх концентрація є важливим нормативним фактором, що впливає на інтенсивність використання глюкози в м'язах.
І, по-друге, жирова тканина також є найважливішим ендокринним органом, який секретує велику кількість чинників, які називаються адипоцитокінами, які мають або сенсиційну дію на інсулін (це адипонектин і лептину), зокрема, фактор некрозу пухлини - альфа (TNF-alpha), резин і т.д.
При надмірній кількості жирових тканин відбувається їх надлишок ліполізу. Зазвичай виділення ФФА від жирових тканин суворо регулюється, що забезпечує інші тканини з чітко збалансованою кількістю ФФА, необхідно адекватно задовольняти свої енергетичні потреби.
Але з ожирінням, патологічно збільшена кількість молекул сигналізації (особливо TNF-alpha) ввести кровоплин, що призводить до порушення метаболічної гомеостазу. Таким чином, ранні події, що призводять до порушення механізму дії інсуліну і виникнення РР, відбуваються в жирових клітинах і довга до настання порушення толерантності до глюкози.
412180 р.
Профіль вільних жирних кислот в сироватці варіюється в залежності від сексу і віку, дієти, зміни гормонального статусу; наприклад, адреналізми або кастинги призводять до зміни РК-профілю.
Зміна концентрації вільного РК, в свою чергу, призводить до значних змін на двох стадіях гормональної передачі інформації: зв'язування глюкокортикоїдів до їх специфічних протеїнів носіїв в плазмі та тканинних рецепторів.
Вільні жирні кислоти більше не вважаються пасивними субстратами, що беруть участь в обмінних процесах. Безкоштовні жирні кислоти є важливими метаболічними сигналами і учасниками ліпідних порушень. У деяких ситуаціях вони полягають як молекули гормонів, які впливають на транскрипцію гена, прив'язуючи до ряду рецепторів.
Серед рецепторів, які зв’язуються з ФФА або їх похідними, спеціальне місце займає окислий проліфератор-активований рецептори – ПАР. Також рецептор G-protein-conjugated GPR120, який виражається в кишечнику, функції як рецептор для ненасичених довгоходових вільних жирних кислот (FFA). Стимуляція GPR120 з вільними жирними кислотами сприяє секреції GLP-1 і збільшує кількість циркуляції інсуліну.
Безкоштовні жирні кислоти і здоров'я.
У зарубіжній медичній літературі, вільні жирні кислоти (FFA) або нестерифіковані жирні кислоти, які утворюються в результаті гідролізу тригліцеридів, що містяться в жирових тканинах, називаються «червоною лампою на тишці міокарда».
Збільшення рівня плазми сигналізує підвищену небезпеку: перший, метаболічний синдром, потім інсулінорезистентність, діабетична кардіоміопатія, а потім ішемічна хвороба серця. Далі ця лампочка може вигорнути, а «світлові цибулини» маркерів некрозу міокардіальної некрозу спалахне, що вказує на те, що «точність не повертається».
Таким чином, підвищені рівні ФФА безпосередньо пов'язані з ожирінням, тобто «прозорі лампочки починають довго світитися до кінцевої точки розвивається», що дозволяє ефективно проводити правильні заходи.
Неймовірні останні дослідження свідчать, що підвищені рівні ФФА, викликані зайвою жировою тканиною, якщо не перший, принаймні один з основних причин ІР. Багато пацієнтів, які страждають ожирінням, метаболічним синдромом і цукровим діабетом II типу, мають підвищені рівні ФФА, що призводить до ІР багатьох тканин - жир, м'яз, печінка, а також ендотеліальні клітини.
FFA - незалежний предиктор нестерпної толерантності до глюкози та DM2. Отже, збільшений потік ФФА від великої маси жирових клітин, а також порушень механізмів, зберігання тригліцеридів і в механізмах ліполізу в тканинах спочатку чутливий до інсуліну – це здається, найбільш ранні прояви аномалії, що призводять до ІР.
Збалансовано, ці порушення виявляються навіть перед розвитком післяпринової гіперглікемії або перед розвитком швидкісної гіперглікемії. Дійсно, збільшення швидкостей плазмових рівнів ФФА, можливо, найбільш ранній показник майбутньої нестерпної толерантності до глюкози.
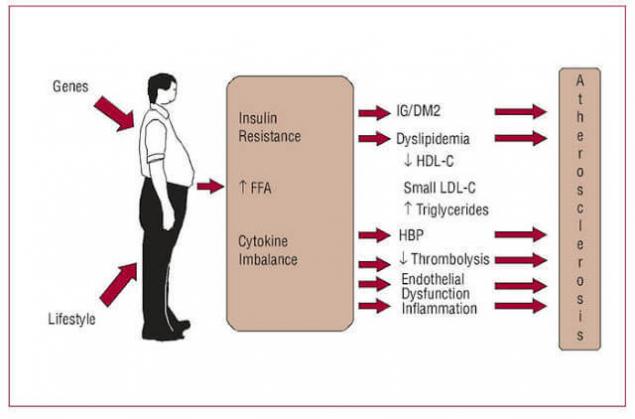
Ізулін опір сходи.
Крок один: порушення регулювання лептину (резистентність лептину).
Розподіл жирних кислот в організмі людини передбачає переважно два гормони: гормон росту, який контролює мобілізацію жирних кислот від жирової тканини, і лептину, що контролює β-окислення жирних кислот в мітохондрії.
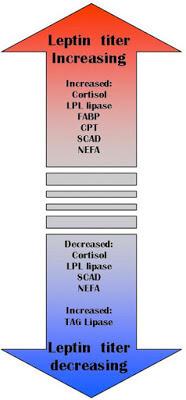
Одним з важливих функцій лептину є збереження тригліцеридів у атипоцитах. Нормальні рівні лептину захищають інші органи від накопичення жиру (веселі, печінка, м'язи тощо). Leptin активує карнітин-палмітоіл-трансферний zu-1, який зв'язує жирну кислоту до карнітину, а останні переносить її через мембрану мітохондріалу, і цей процес суворо регулюється. Лептин також стимулює окислення жирних кислот і зменшує кількість триацилгліцеролів в м'язовій тканині. Лептин також був знайдений для гальмування активності ацетил-КоА карбоксильази!
Хронічний стрес, перекриття, неправильне харчування, надлишок цукру, гіподинамія призведе до порушення системи лептину. Якщо опір лептину виникає, це призводить до збільшення кількості вільних жирних кислот. Другий крок - хронічне збільшення ФФА.
Крок два: збільшення в'язкої жирової тканини.
Це в'язальна жирова тканина, яка стане найпотужнішим джерелом ФФА. В'язкість жиру підвищується пропорційно індексу маси тіла і є незалежним предикатором цукрового діабету 2 типу. В'язкова жирова тканина є основним джерелом вільних жирних кислот (FFA).
З вісцеральним ожирінням зайва кількість вільних жирних кислот надходить в печінку через портал вену систему ( 20-30 разів вище норми), яка виводить печінку на серйозні перевантаження і, в результаті призводить до розвитку вищезазначених порушень обміну речовин.
В’язковий жир, що присутній навколо внутрішніх органів, мезентерії та оменту, відрізняється від підшкірного жиру в типі атипоцитів, їх ендокринна функція, ліполітична активність, чутливість до інсуліну та інших гормонів.
На відміну від підшкірної жирової тканини, венозний кровоплин від в'язкої маси через порталну систему безпосередньо надходить в печінку. Це викликає безпосереднє надходження великої кількості вільних жирних кислот (FFA) і адсипокінів в печінку, синтезованих у в'язкій жировій тканині.
Адіпокини, в свою чергу, активують імунні механізми печінки, що призводять до утворення прозапальних медіаторів, таких як C-реактивний білок (CRP) та ін. Безкоштовні жирні кислоти, в великих кількостях, що надходять в печінку з вісцеральної жирової тканини, викликають розвиток печінкової інсулінорезистентності.
За секретом моноциклічної хіміоатрактантної білки-1 (MCP-1), яка сприяє інфільтрації макрофагів жирової тканини, адипоцитів викликає прозапальний стан.
Macrophages, в свою чергу, є важливим джерелом прозапальних цитокінів, таких як фактор некрозу пухлини-α (TNF-α) і interleukin-6 (IL-6). В'язкова жирова тканина більш інфільтрована запальними клітинами, і тому секрети великої кількості прозапальних цитокінів у порівнянні з підшкірним жиром.
Етопічний жир - це, жир, який не в підшкірному жирі. Цей жир найчастіше вісцеральний, або печінковий, або міжм'язовий. Але це об’єднує той факт, що він не підшкірний жир, але знаходиться на місці «вороги». Подальший розвиток захворювання буде залежати від того, які тканини, які не призначені для їх зберігання, будуть накопичуватися ФФА. Якщо накопичуються в скелетних м'язах - це призведе до ІР, якщо в печінці - до дисліпідемії. По-перше, як правило, ІЧ розвивається, потім, з його ваговим, ішемічною хворобою серця - ІБС.
Крок три: хронічне збільшення ФФА.
Як показано вище, при в'язкому ожиріння, зайва кількість вільних жирних кислот надходить в печінку через портал вену систему ( 20-30 разів вище норми), яка виводить печінку на серйозні перевантаження і, в результаті призводить до розвитку вищенаведених метаболічних порушень. У крові хронічний підвищений рівень ФФА. Разом з лептинною стійкістю, це поступово призводить до збільшення кількості жиру в нежирних органах.
Під дією ФФА в жировій тканині утворюються більші адсипоцити, які стійкі до дії інсуліну, процес локального запалення ініціюється секреція прозапальних цитокінів.
Хронічне підвищення рівня жирних кислот в крові є наслідком порушення в організмі системи регулювання їх гомеостазу. Стійкість до лептину, що дозволяє запобігти надлишку деяких його стаціонарних лімітів окислення і, отже, утилізація надлишкових жирних кислот в мітохондрії.
Таким чином, як ми можемо припустити, виникає ситуація, коли, через підвищений вміст жирних кислот в периклітинній трансфузії, їх потік в клітинку підвищується (за рахунок збільшення вмісту жирних кислот в організмі через їх примирення), а при одночасному резистентності до лептину, окислення жирних кислот залишається на одному рівні.
Ймовірно, відгуки між надходженням жирних кислот в клітинку нежирної тканини і секретацією їх в кров або зламаний, або не існує взагалі, тобто немає механізму підтримки стаціонарного рівня жирних кислот в крові. Якщо це припущення є справжньою, то в цьому відношенні регуляція жирового метаболізму кислоти принципово відрізняється від регулювання метаболізму глюкози, стаціонарний рівень якого підтримується гормоновою системою.
Під контролем, ймовірно, тільки окислення жирних кислот в мітохондрії, тобто внутрішньоклітинне використання цього енергетичного субстрату. В останні роки досліджено механізми регулювання потоку жирних кислот в організмі.
Відкрито сімейство ядерних рецепторів ПАР, вони стали відомими завдяки здатності викликати проліферацію оксімії і карциногенез в печінці у відповідь на ксенобіотиків. Виявлено три ізоформи PPAR, α, γ та δ, а також властивості рецептів PPARα та PPARγ. Ліганди для рецепторів насичені, ненасичені і мононенасичені жирні кислоти.
PPARγ виражається при адипоцитах і зменшує секрецію жирних кислот в кров від жирової тканини. PPARα виражається в клітинах печінки, скелетних і серцевих м'язів і виступає як «ліпотат», що регулює процеси внутрішньоклітинного синтезу і β-окислення жирних кислот в мітохондрії і оксіомах.
ППАРи стимулюються лептином, гормоном росту і інсуліном, їх експресія заперечує ритм циркадії, вони виражені у відповідь на споживання їжі. Ці рецептори здійснюють внутрішньоклітинне регулювання жирних кислот, зберігаючи стаціонарний рівень споживання енергії клітиною, але вони не здаються залученню до утримання гомеостазу жирних кислот на рівні тіла.
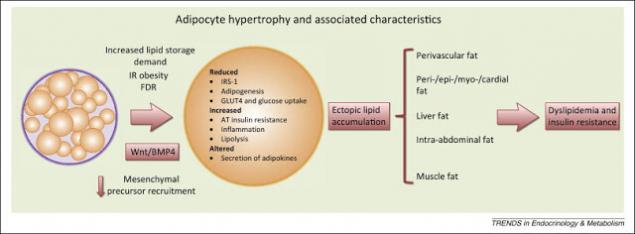 р.
р.
Крок четвертий: Виникнення образу.
Збільшення рівня ФФА призводить до їх накопичення в клітинах, реструктуризації клітинних мембран і зменшення інсулінорезистентності. Надлишок тригліцеридів у клітинах викликає збільшення синтезу запальних цитокінів. Це жирова тканина, яка в даний час розглядається як місце початкового виникнення і розвитку ІР. Це пов'язано з: (а) підвищеними рівнями ФФА в кровоплину і, (2) підвищеною секрецією адипоцитокінів.
Велику масу адипоцитів синтетифікує збільшення кількості прозапальних цитокінів, що призводить до хронічного запального процесу, що: (a) порушує хід по сигналізації інсуліну та (b) пошкодження мітохондріальної функції, яка порушує гомілкозний гомеостаз. Зокрема, IL-6 і TNF-альфа, що виділяються жировими клітинами, що посилюють IR, і секретують ангіотензин II підвищує артеріальний тиск і сприяє розвитку атеросклерозу.
Порушення адаптивного механізму.
У клітині жирних кислот не споживають для β-окислення, фосфоліпіди спочатку синтезуються, а потім тригліцериди, які накопичуються в цитоплазмі. внутрішньоклітинні тригліцериди в нежирних тканинах містять переважно пальмову кислоту. З пальмової кислоти, sphingomyelin синтезується, яка є основною складовою мембранних рафонів, що беруть участь у регулюванні активності мембранних рецепторів.
Синтез сфінгомієліну, який залежить від вмісту пальмової кислоти в клітині, здійснюється по шляху «пальмітова кислота → керамід → сфінгомієлін». Це спосіб синтезу кераміду з пальмової кислоти, що призводить до окислювального апоптозу. Церамід – це індуктор апоптозу як вздовж окислювального шляху (керамід блокує комплекс ІІІ ЕТК, що викликає збільшення окислювачів), так і без залучення мітохондрій.
Скупчення тригліцеридів в кардіоміоцитах пов'язана з зменшенням синтезу кардіоліпину і зміною дихальної функції мітохондрії, так як цитохром з окислотою комплексу IV ETS пов'язаний з кардіоліпином. Зміна структури мітохондріальної мембрани призводить до виходу цитохрому c і апоптозу без участі окислювачів. Таким чином, накопичення пальмової кислоти в клітинах нежирних тканин призводить до збільшення синтезу кераміду і зменшення синтезу кардіоліпину, що викликає апоптоз, і до зміни активності рецепторів.
У зв'язку з накопиченням тригліцеридів у клітинах (тригліцериди самі не викликають апоптозу) вважається спробою тіла, щоб уникнути ефекту ліпотоксичності.
Сфінгомієлін і пальмова кислота виявляє високу афінність для холестерину. Підвищення вмісту сфінгомієліну та пальмової кислоти в мембрані може пояснити вікове накопичення холестерину в мембранах, а також зміна чутливості рецептора інсуліну.
4961.31
Інсулінний рецептор асоціюється з мембранними рафтингами, а зміна складу рафтів впливає на його чутливість. Скупчення тригліцеридів у нежирних тканинах і пов'язаного зниження чутливості рецептора інсуліну призводить до виникнення інсулінорезистентності і гіперглікемії, тобто підвищеного вмісту глюкози в крові. Інсулін рецептор – кінази тирозин.
За допомогою автофосфориляції активуються різні доріжки, зокрема, PI-3-K шляхової дороги (фосінозитол-3-кенази), через які відбувається транспортування глюкози в клітинку, оскільки транспортер глюкози GLUT4 входить до його активного робочого стану.
У зв'язку з активним ліполізом вільних жирних кислот (FFA) і прозапальних цитокінів, вони впливають на субстрати рецептора інсуліну і тим самим блокують PI-3-K шляховий шлях, в результаті чого блокуються ефекти, що цей шлях має на метаболізмі глюкози, і глюкоза не може ввести клітинку. Таким чином, інсулінорезистентність розвивається, тобто надмірна кількість в'язкої жирової тканини блокує вхідний сигнал і призводить до того, що інсулінові рецептори стають нечутливими до інсуліну, а її біологічна роль спотворюється.
Залежно від індивідуальної чутливості та генетичності, інсулінорезистентність може розвиватися в різних тканинах. Надмірне ФФА повідомляє про прогресування інсулінорезистентності багатьох тканин - м'язів, включаючи міокард, гепатичний, адсипоз, а також ендотеліальні клітини, сприяє прогресуванню ішемічних змін в міокарда, включаючи зміни, пов'язані з порушенням бета-окислення ФФА в міокарда.
Крок п'ять: печінковий і м'язовий інсулінорезистентність.
В умовах підвищеного надходження їжі, багатих жирами і вуглеводами, стимулюється секреція інсуліну, яка, в свою чергу, активізує ліпогенез і відкладення ФФА в жировій тканині. Однак генетично визначається обмеження здатності накопичувати ліпіди, тому при об'ємі жирової тканини досягає максимального, надлишок ФФА починає входити в печінку і м'язи.
Надлишок ФФА супроводжується накопиченням тригліцеридів в пароенхімальних клітинах багатьох тканин, а саме в скелетних і кардіоміоцитах і в гепатоцитах, що призводить до їх пошкодження і хронічної дисфункції.
В результаті печінка в умовах інсулінорезистентності починає активно синтезувати жирні кислоти, тригліцериди, ліполіз прискорює, але вже в жировій тканині. Крім того, всі ті процеси виникають в печінці, які пригнічують пацієнта з вісцеральним ожирінням до цукрового діабету: глюкозогенез стимулюється і пригнічують гліколіз і синтез глікогену.
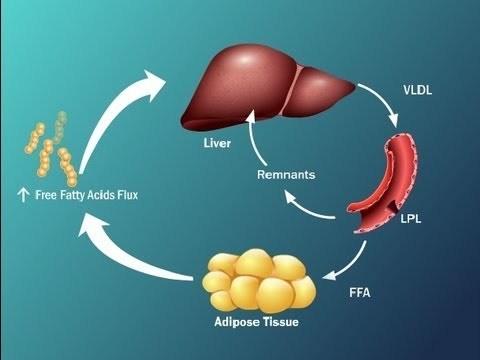
Досить складна схема виявлена, що відображає, що інсулінорезистентність, або інсулінорезистентність, призводить до того, що печінка перевантажується жирними кислотами.
Це пов'язано з тим, що активні жирні кислоти синтезуються в печінці, окислення жирних кислот знижується, жирні кислоти активно йдуть в печінку з вісцеральної жирової тканини і, крім того, жир, який надходить в нашу печінку в складі хіміомікронів, також перевантаження печінки з вільними жирними кислотами.
Ці процеси призводять до того, що печінка не здатна метаболізувати β-окислення ФФА, перекислення ліпідів відбувається, в результаті чого утворюються реактивні види кисню в великих кількостях, виникає окислювальний стрес, і це такі фактори, що призводять до фосфонії інсулінопроцедурної субстрату, які ми говорили про в попередньому слайді, тим самим, інсулінорезистентність знову викликається, тобто різновид замкненого кола, і вже важко визначити у пацієнта, що є первинним.
Крім того, доведено, що макрофаги в'язкої жирової тканини мають прозапальну активність. Крім того, CD-8 + Т-лімфоцити знаходяться в жировій тканині, яка активно секретує прозапальні цитокіни і, таким чином, в печінці в стані жирової дистрофії вже може переходити на наступний етап з розвитком NASH у хворих та інших наслідків, до яких ведеться NAFLD.
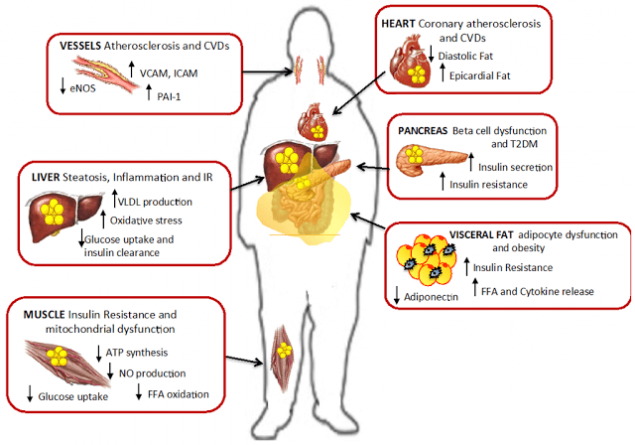
М'язова інсулінорезистентність.
Незабаром після печінки жир починає накопичуватися в м'язах. метаболічна патологія, характерна для ІР, є накопичення тригліцеридів навколо м'язових фібрилів. Однак скупчення тригліцеридів всередині скелетних м'язів не вважається безпосередньою причиною розвитку цукрового діабету 2, але, здається, є маркером ліпідних проміжних сполук, таких як ацетил КоА, кераміди і діакілгліцерин.
За останніми дослідженнями порушення шляхів передачі інсуліну в основному пов'язана з патологічним обміном ФФА в скелетних м'язових клітинах, які «не впоратися» з їх використанням при перевищенні ФФА. Дійсно, локальне накопичення всередині скелетних м'язів таких метаболітів ФФА як кераміди, діагліцерол або ацил-КоА призводить до порушення передавання інсулінових сигналів і, тим самим, до порушення глюкози транспорту.
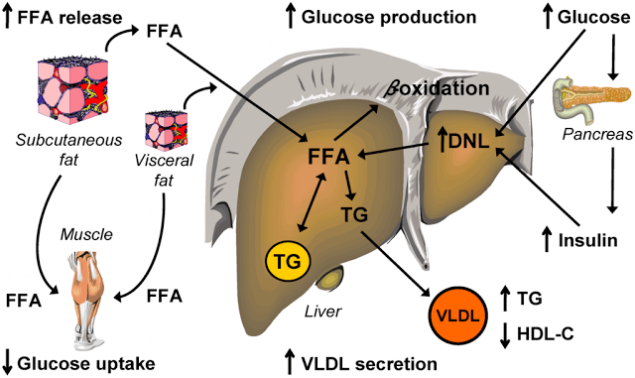
Крок шість. Злий цикл інсулінорезистентності.
інсулінорезистентність, викликана високими рівнями ФФА, додатково збільшує концентрацію ФФА в плазмі. Як знайдено, інсулінорезистентні жирові клітини секрети підвищених рівнів ФФА. Це, фактично, дозволяє розглянути підвищені рівні ФФА як маркер ІР.
Дійсно, з ІР рівень ФФА в гепатоцитах зростає, тому що в них:
(1) підвищується ново-ліпогенез,
(2) естерифікація ФФА перевищує їх окислення,
(3) естерифіковані РК-дисплеї зберігаються як тригліцериди або відправлені для синтезу VLDL-X (багатих тригліцеридів),
При зменшенні інсулін-регульованої тригліцеридної мобілізації.
Інсуліностійкі асоціоцити інтенсивно порушують тригліцериди, що містяться в них і випускають ФФА, утворену з них в кровотік (як з ожирінням і без нього). Потік ФФА з жирових клітин підвищується і, крім того, ФФА також залишає X-VLDL і з плазмохімічних показників і через кровоплин частково спрямований на інші органи, а частково - назад до печінки, де вони знову перетворюються в тригліцериди. Є «пампінг» печінки ФФА і тригліцеридів. Це має найбільш серйозні наслідки.
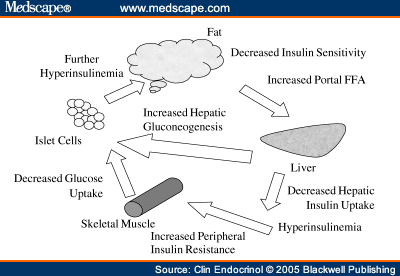
Крок 7. Прискорений атеросклероз.
Підвищені рівні ФФА призводять до дисліпідемії та атерогенезу
Як інсулінорезистентність призводить до дисліпідемії Підвищені тригліцериди печінки стимулюють утворення Apo B і X-VLDL,
Ось як це відбувається.
1 час З печінки високі рівні VLDL-X виділяються в плазму, де ліполіз від VLDL-X утворює FFA і високоатеросогенний залишок (відмова) частинок тригліцерид-багатих ліпопротеїнів.
1 час Від плазми, FFA і залишків частинок знову поглинаються печінкою, яка додатково збільшує рівень ФФА в гепатоцитах і додатково стимулює синтез X-VLDL.
(1) У печінці з високим рівнем VLDL-X і нормальними рівнями білка CETP (холестериловий протеїн) - носієм холестерину ефіру, тригліцеридів від VLDL-X переходить в HDL-X, а холестерин від HDL-X переходить в VLDL-X. В результаті: (a) холестерин-багатий дуже атеросогенний залишок X-VLDL частинок і (b) HDL X, що містить багато тригліцеридів і невеликий холестерин.
(4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) Такі частинки HDL-X втрачають тригліцериди (під впливом печінкової губи) і їх головна аполіпопротеїн Apo A1. В результаті знижується рівень антитерогенного X-HDL.
(5) (4) (5) (1) (5) (1) (5) (1) (5) (1) (5) (1) (5) (1) (5) (1) (5) (1) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (4) (5) (4) (5) (4) (5) (5) (4) (5) (4) (5) (5) (4) (5) (4) (5) (4) (5) (5) (4) (5) (4) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) На високих рівнях VLDL-X (багаті тригліцериди), CETP несе тригліцериди від VLDL-X до LDL-X і холестерину від LDL-X до VLDL-X.
(6) Багаті тригліцериди X-LDL через активність печінкових або ліпопротеїнових ліпідів втрачають тригліцериди, зменшують розмір і стають дуже атеросогенними невеликими щільні частинки X-LDL.
Таким чином, підвищені рівні ФФА призводять до зменшення рівня «антиатерогенної» HDL-X, утворення вкрай атеросогенних дрібних, щільних LDL-C частинок і підвищення рівня тригліцериду плазми.
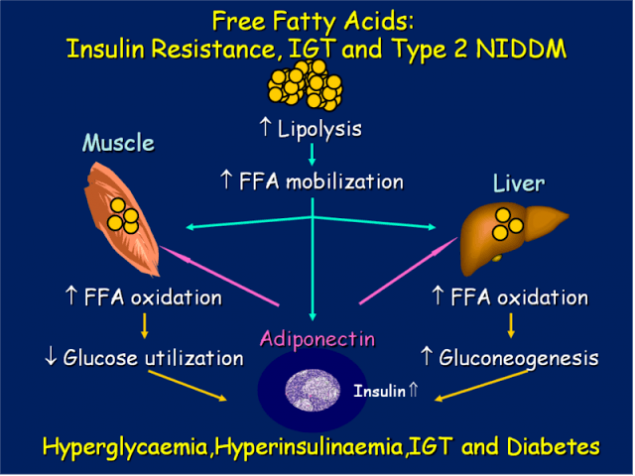
Але є ще один спосіб, що високий рівень FFA викликає атерогенез. Цей шлях є більш прямим і коротким. Підвищений рівень ФФА в ІР викликає в мітохондрії макросудинних ендотеліальних клітин надсинтез реактивних видів кисню, що призводить до окислення ЛЛ-Х і модифікації HDL-X. Це викликає запальний процес в стінках судин, утворення і накопичення холестеринових нальотів і, в результаті ішемії. Видання
Джерела.
1. Публікації В.В. Велкова
2. В.В. Велков. Діагностика праці. - 2009. - Т. 3, No 49. - С. 55-71.
3. У Вельков В.В. Вільні жирні кислоти – фактор ризику інсулінорезистентності та ішемії: перспективи оцінки ризику та діагностики
4. Л.І. Данилова феномен інсулінорезистентності в клінічній практиці: механізми формування та можливості корекції
5. Умань Е.В. Терешина Роль жирних кислот у розвитку вікового окислювального стресу. Пороги в геронології, 2007. Т. 20, No 1
6. Ендокринологія та метаболізм, Ф. Феліг, Д. Бакстер
Автор: Андрій Білловкін
Психосоматика захворювань шиї: Через наші горлі ми ковтати реальність.
Що не варто лікувати
Джерело: www.beloveshkin.com/2016/01/svobodnye-zhirnye-kisloty-klyuch-k-ponimaniyu-insulinorezistentnosti.html
Тому сьогодні я буду говорити про FFA - безкоштовні жирні кислоти (у ілюстраціях, позначених FFA - безкоштовні жирні кислоти). З одного боку, FFA є основним джерелом енергії, але з іншого боку, це важлива молекула сигналізації.
Я особливо хочу відзначити, що коли мова йде про хронічне збільшення ФФА як причину інсулінорезистентності (ІР), то пам'ятайте, що проблема не ФФА, але що вони ніде використовувати, тому їх надлишки з'являються в крові. Ця стаття є довгою, але хто цікавиться розумінням логіки здоров'я та хвороби.

Безкоштовні жирні кислоти: Ключ до розуміння
Безкоштовні жирні кислоти.
В результаті гідролізу тригліцеридів, що містяться в жирових тканинах. Плазма жирні кислоти є естерифікованими і в цьому випадку є переважно альбумінними або не естерифікованими і безкоштовні. Безкоштовні жирні кислоти (FFA) є частиною 5 ліпопротеїнів.
Останній складається з довгоочіданих жирних кислот, міцно зв'язаних з двома певними регіонами молекули альбуміну; при збільшенні рівня ФФА в плазмі вони займають додаткові ділянки цієї молекули, але зв'язування в цьому випадку менш сильний.
FFAs є основною енергетичною субстратністю тіла. Вони утворюються в процесі ліполізу тригліцеридів, накопичених в клітинах жирової тканини. Ліза тканин в цих клітинах під нейроендокринним контролем і активується через систему циклази аденілат.
Друге джерело плазми FFA є гідролізом тригліцеридів, що містяться в ліпопротеїнів, під впливом ліпопротеїну ліпоза. Уміння скелетних м’язів (та інших тканин) для регулювання їх метаболізму до домінантою субстрату в даний час зазвичай називають «добрим метаболічним здоров’ям» або «метаболічною гнучкістю». Зрозуміло, що хороше «метаболічне здоров’я» пов’язане з нормальною чутливістю до інсуліну.
Безкоштовний обмін жирною кислотою.
Період напіврозпаду ФФА дуже короткий - 4 - 8 хвилин, і вони легко поглинаються з плазми м'язовими клітинами тіла. Другим способом їх обміну є всмоктування печінки і ресинтезу в тригліцериди, які потім можна перевозити з печінки в складі VLDL або окислюється до ацетил-CoA. У фізіологічних умовах рівень ФФА в крові може швидко зростатися і знизитися, наповнювати потреби організму для цієї форми енергії.
Вміст їх зазвичай нижчий після поглинання вуглеводів і отриманого випуску інсуліну, але в міру зниження рівня глюкози в крові після їжі їх рівень збільшується. На порожньому шлунку крові міститься, як правило, 400 - 600 мкЕк / л ФФА; з більшою швидкістю (до 24 - 72 годин), рівень ФФА може досягати 1000 - 1500 мкЕк / л. Глюкагон, адреналін, гормону росту і АКТ також підвищують рівень ФФА. Основними фізіологічними регуляторами вмісту ФФА в плазмі є інсулін і адреналін.
Кожна хвилина, 20-40% ФФА, що надходить до плазми, вводяться окислені, рестеризовані або перетворюються на інші жирні кислоти. Під час відпочинку окислення відбувається переважно в печінці і серці, а під час стресу - в скелетних м'язах, а в останньому випадку частка окислюється ФФА збільшується від близько 20 до 60%.
Більшість ФФА, захоплених клітинами печінки, переоцінюються для формування переважно тригліцеридів, а також фосфоліпідів, для синтезу яких зазвичай використовується лінолеєва кислота. У плазмі FFAs міститься в діапазоні концентрацій від 100 мкмоль / л до 1 ммоль / л і їх рівень сильно залежить від часу доби.
Після кожної їжі рівень ФФА в плазмі осені, так як інсулін пригнічує ліполіз у жирових клітинах, в результаті чого ФФА утворюється. На ніч збільшується концентрація ФФА в плазмі. Для цих нормальних щоденних коливань в рівні ФФА практично всіх інших тканин, зокрема, скелетних м'язів, «свербіж» від використання глюкози (день) до споживання ФФА (нині).
Деякі причини порушення обміну ФФА.
Ключовим порушенням метаболізму вільних жирних кислот є їх хронічне збільшення. Хронічне висіння рівнів ФФА може бути викликане різними причинами харчової та стрес-типу. Наприклад, надлишок вуглеводів, в яких починається синтез нових жирних кислот від зайвих вуглеводів.
З 75-80% глюкози у здоровій людини вкладається скелетний м'яз, фізична активність робітників може запобігти розвитку «функціональної» інсулінорезистентності. Під час фізичного навантаження також викликає збільшення ФФА.
Хронічний стрес будь-якого типу є ще однією причиною хронічного збільшення ФФА. Під час стресу в крові рівень ФФА підвищується, щоб забезпечити роботу серця і м'язів. Але людина зазвичай не рухається і всі ці жирні кислоти циркулюють в крові протягом тривалого часу.
Великомасштабними дослідженнями стали неперервні зв’язки між становищем людини на соціально-економічній сході та ймовірність розвитку метаболічного синдрому. Авторами висновку, що розвиток метаболічного синдрому є біологічним механізмом, що призводить до «соціальної нерівності в розподілі коронарного ризику серед людей. й
Високий коронарний ризик (завищені рівні тригліцериду і низькі рівні холестерину ліпопротеїну високої щільності) пов'язані з низьким соціально-економічним статусом (дорослим), що в дорослому віці призводить до зайвої ваги і ожиріння.
Крім того, психологічний стрес також сприяє розвитку T1D і T2D, так як високі рівні ФФА асоціюються з підвищеним утворенням в мітохондрії реактивних видів кисню (окислювальний стрес), що в кінцевому рахунку призводить до зменшення здатності клітин реагувати на адекватно дії інсуліну.
FFA - це метаболіт і молекула сигналізації.
Чисельні дослідження в останні роки показали, що, по-перше, FFA не тільки високоенергетичне паливо, але й важливі молекули сигналізації. Їх концентрація є важливим нормативним фактором, що впливає на інтенсивність використання глюкози в м'язах.
І, по-друге, жирова тканина також є найважливішим ендокринним органом, який секретує велику кількість чинників, які називаються адипоцитокінами, які мають або сенсиційну дію на інсулін (це адипонектин і лептину), зокрема, фактор некрозу пухлини - альфа (TNF-alpha), резин і т.д.
При надмірній кількості жирових тканин відбувається їх надлишок ліполізу. Зазвичай виділення ФФА від жирових тканин суворо регулюється, що забезпечує інші тканини з чітко збалансованою кількістю ФФА, необхідно адекватно задовольняти свої енергетичні потреби.
Але з ожирінням, патологічно збільшена кількість молекул сигналізації (особливо TNF-alpha) ввести кровоплин, що призводить до порушення метаболічної гомеостазу. Таким чином, ранні події, що призводять до порушення механізму дії інсуліну і виникнення РР, відбуваються в жирових клітинах і довга до настання порушення толерантності до глюкози.
412180 р.
Профіль вільних жирних кислот в сироватці варіюється в залежності від сексу і віку, дієти, зміни гормонального статусу; наприклад, адреналізми або кастинги призводять до зміни РК-профілю.
Зміна концентрації вільного РК, в свою чергу, призводить до значних змін на двох стадіях гормональної передачі інформації: зв'язування глюкокортикоїдів до їх специфічних протеїнів носіїв в плазмі та тканинних рецепторів.
Вільні жирні кислоти більше не вважаються пасивними субстратами, що беруть участь в обмінних процесах. Безкоштовні жирні кислоти є важливими метаболічними сигналами і учасниками ліпідних порушень. У деяких ситуаціях вони полягають як молекули гормонів, які впливають на транскрипцію гена, прив'язуючи до ряду рецепторів.
Серед рецепторів, які зв’язуються з ФФА або їх похідними, спеціальне місце займає окислий проліфератор-активований рецептори – ПАР. Також рецептор G-protein-conjugated GPR120, який виражається в кишечнику, функції як рецептор для ненасичених довгоходових вільних жирних кислот (FFA). Стимуляція GPR120 з вільними жирними кислотами сприяє секреції GLP-1 і збільшує кількість циркуляції інсуліну.
Безкоштовні жирні кислоти і здоров'я.
У зарубіжній медичній літературі, вільні жирні кислоти (FFA) або нестерифіковані жирні кислоти, які утворюються в результаті гідролізу тригліцеридів, що містяться в жирових тканинах, називаються «червоною лампою на тишці міокарда».
Збільшення рівня плазми сигналізує підвищену небезпеку: перший, метаболічний синдром, потім інсулінорезистентність, діабетична кардіоміопатія, а потім ішемічна хвороба серця. Далі ця лампочка може вигорнути, а «світлові цибулини» маркерів некрозу міокардіальної некрозу спалахне, що вказує на те, що «точність не повертається».
Таким чином, підвищені рівні ФФА безпосередньо пов'язані з ожирінням, тобто «прозорі лампочки починають довго світитися до кінцевої точки розвивається», що дозволяє ефективно проводити правильні заходи.
Неймовірні останні дослідження свідчать, що підвищені рівні ФФА, викликані зайвою жировою тканиною, якщо не перший, принаймні один з основних причин ІР. Багато пацієнтів, які страждають ожирінням, метаболічним синдромом і цукровим діабетом II типу, мають підвищені рівні ФФА, що призводить до ІР багатьох тканин - жир, м'яз, печінка, а також ендотеліальні клітини.
FFA - незалежний предиктор нестерпної толерантності до глюкози та DM2. Отже, збільшений потік ФФА від великої маси жирових клітин, а також порушень механізмів, зберігання тригліцеридів і в механізмах ліполізу в тканинах спочатку чутливий до інсуліну – це здається, найбільш ранні прояви аномалії, що призводять до ІР.
Збалансовано, ці порушення виявляються навіть перед розвитком післяпринової гіперглікемії або перед розвитком швидкісної гіперглікемії. Дійсно, збільшення швидкостей плазмових рівнів ФФА, можливо, найбільш ранній показник майбутньої нестерпної толерантності до глюкози.

Ізулін опір сходи.
Крок один: порушення регулювання лептину (резистентність лептину).
Розподіл жирних кислот в організмі людини передбачає переважно два гормони: гормон росту, який контролює мобілізацію жирних кислот від жирової тканини, і лептину, що контролює β-окислення жирних кислот в мітохондрії.

Одним з важливих функцій лептину є збереження тригліцеридів у атипоцитах. Нормальні рівні лептину захищають інші органи від накопичення жиру (веселі, печінка, м'язи тощо). Leptin активує карнітин-палмітоіл-трансферний zu-1, який зв'язує жирну кислоту до карнітину, а останні переносить її через мембрану мітохондріалу, і цей процес суворо регулюється. Лептин також стимулює окислення жирних кислот і зменшує кількість триацилгліцеролів в м'язовій тканині. Лептин також був знайдений для гальмування активності ацетил-КоА карбоксильази!
Хронічний стрес, перекриття, неправильне харчування, надлишок цукру, гіподинамія призведе до порушення системи лептину. Якщо опір лептину виникає, це призводить до збільшення кількості вільних жирних кислот. Другий крок - хронічне збільшення ФФА.
Крок два: збільшення в'язкої жирової тканини.
Це в'язальна жирова тканина, яка стане найпотужнішим джерелом ФФА. В'язкість жиру підвищується пропорційно індексу маси тіла і є незалежним предикатором цукрового діабету 2 типу. В'язкова жирова тканина є основним джерелом вільних жирних кислот (FFA).
З вісцеральним ожирінням зайва кількість вільних жирних кислот надходить в печінку через портал вену систему ( 20-30 разів вище норми), яка виводить печінку на серйозні перевантаження і, в результаті призводить до розвитку вищезазначених порушень обміну речовин.
В’язковий жир, що присутній навколо внутрішніх органів, мезентерії та оменту, відрізняється від підшкірного жиру в типі атипоцитів, їх ендокринна функція, ліполітична активність, чутливість до інсуліну та інших гормонів.
На відміну від підшкірної жирової тканини, венозний кровоплин від в'язкої маси через порталну систему безпосередньо надходить в печінку. Це викликає безпосереднє надходження великої кількості вільних жирних кислот (FFA) і адсипокінів в печінку, синтезованих у в'язкій жировій тканині.
Адіпокини, в свою чергу, активують імунні механізми печінки, що призводять до утворення прозапальних медіаторів, таких як C-реактивний білок (CRP) та ін. Безкоштовні жирні кислоти, в великих кількостях, що надходять в печінку з вісцеральної жирової тканини, викликають розвиток печінкової інсулінорезистентності.
За секретом моноциклічної хіміоатрактантної білки-1 (MCP-1), яка сприяє інфільтрації макрофагів жирової тканини, адипоцитів викликає прозапальний стан.
Macrophages, в свою чергу, є важливим джерелом прозапальних цитокінів, таких як фактор некрозу пухлини-α (TNF-α) і interleukin-6 (IL-6). В'язкова жирова тканина більш інфільтрована запальними клітинами, і тому секрети великої кількості прозапальних цитокінів у порівнянні з підшкірним жиром.
Етопічний жир - це, жир, який не в підшкірному жирі. Цей жир найчастіше вісцеральний, або печінковий, або міжм'язовий. Але це об’єднує той факт, що він не підшкірний жир, але знаходиться на місці «вороги». Подальший розвиток захворювання буде залежати від того, які тканини, які не призначені для їх зберігання, будуть накопичуватися ФФА. Якщо накопичуються в скелетних м'язах - це призведе до ІР, якщо в печінці - до дисліпідемії. По-перше, як правило, ІЧ розвивається, потім, з його ваговим, ішемічною хворобою серця - ІБС.
Крок три: хронічне збільшення ФФА.
Як показано вище, при в'язкому ожиріння, зайва кількість вільних жирних кислот надходить в печінку через портал вену систему ( 20-30 разів вище норми), яка виводить печінку на серйозні перевантаження і, в результаті призводить до розвитку вищенаведених метаболічних порушень. У крові хронічний підвищений рівень ФФА. Разом з лептинною стійкістю, це поступово призводить до збільшення кількості жиру в нежирних органах.
Під дією ФФА в жировій тканині утворюються більші адсипоцити, які стійкі до дії інсуліну, процес локального запалення ініціюється секреція прозапальних цитокінів.
Хронічне підвищення рівня жирних кислот в крові є наслідком порушення в організмі системи регулювання їх гомеостазу. Стійкість до лептину, що дозволяє запобігти надлишку деяких його стаціонарних лімітів окислення і, отже, утилізація надлишкових жирних кислот в мітохондрії.
Таким чином, як ми можемо припустити, виникає ситуація, коли, через підвищений вміст жирних кислот в периклітинній трансфузії, їх потік в клітинку підвищується (за рахунок збільшення вмісту жирних кислот в організмі через їх примирення), а при одночасному резистентності до лептину, окислення жирних кислот залишається на одному рівні.
Ймовірно, відгуки між надходженням жирних кислот в клітинку нежирної тканини і секретацією їх в кров або зламаний, або не існує взагалі, тобто немає механізму підтримки стаціонарного рівня жирних кислот в крові. Якщо це припущення є справжньою, то в цьому відношенні регуляція жирового метаболізму кислоти принципово відрізняється від регулювання метаболізму глюкози, стаціонарний рівень якого підтримується гормоновою системою.
Під контролем, ймовірно, тільки окислення жирних кислот в мітохондрії, тобто внутрішньоклітинне використання цього енергетичного субстрату. В останні роки досліджено механізми регулювання потоку жирних кислот в організмі.
Відкрито сімейство ядерних рецепторів ПАР, вони стали відомими завдяки здатності викликати проліферацію оксімії і карциногенез в печінці у відповідь на ксенобіотиків. Виявлено три ізоформи PPAR, α, γ та δ, а також властивості рецептів PPARα та PPARγ. Ліганди для рецепторів насичені, ненасичені і мононенасичені жирні кислоти.
PPARγ виражається при адипоцитах і зменшує секрецію жирних кислот в кров від жирової тканини. PPARα виражається в клітинах печінки, скелетних і серцевих м'язів і виступає як «ліпотат», що регулює процеси внутрішньоклітинного синтезу і β-окислення жирних кислот в мітохондрії і оксіомах.
ППАРи стимулюються лептином, гормоном росту і інсуліном, їх експресія заперечує ритм циркадії, вони виражені у відповідь на споживання їжі. Ці рецептори здійснюють внутрішньоклітинне регулювання жирних кислот, зберігаючи стаціонарний рівень споживання енергії клітиною, але вони не здаються залученню до утримання гомеостазу жирних кислот на рівні тіла.
 р.
р.Крок четвертий: Виникнення образу.
Збільшення рівня ФФА призводить до їх накопичення в клітинах, реструктуризації клітинних мембран і зменшення інсулінорезистентності. Надлишок тригліцеридів у клітинах викликає збільшення синтезу запальних цитокінів. Це жирова тканина, яка в даний час розглядається як місце початкового виникнення і розвитку ІР. Це пов'язано з: (а) підвищеними рівнями ФФА в кровоплину і, (2) підвищеною секрецією адипоцитокінів.
Велику масу адипоцитів синтетифікує збільшення кількості прозапальних цитокінів, що призводить до хронічного запального процесу, що: (a) порушує хід по сигналізації інсуліну та (b) пошкодження мітохондріальної функції, яка порушує гомілкозний гомеостаз. Зокрема, IL-6 і TNF-альфа, що виділяються жировими клітинами, що посилюють IR, і секретують ангіотензин II підвищує артеріальний тиск і сприяє розвитку атеросклерозу.
Порушення адаптивного механізму.
У клітині жирних кислот не споживають для β-окислення, фосфоліпіди спочатку синтезуються, а потім тригліцериди, які накопичуються в цитоплазмі. внутрішньоклітинні тригліцериди в нежирних тканинах містять переважно пальмову кислоту. З пальмової кислоти, sphingomyelin синтезується, яка є основною складовою мембранних рафонів, що беруть участь у регулюванні активності мембранних рецепторів.
Синтез сфінгомієліну, який залежить від вмісту пальмової кислоти в клітині, здійснюється по шляху «пальмітова кислота → керамід → сфінгомієлін». Це спосіб синтезу кераміду з пальмової кислоти, що призводить до окислювального апоптозу. Церамід – це індуктор апоптозу як вздовж окислювального шляху (керамід блокує комплекс ІІІ ЕТК, що викликає збільшення окислювачів), так і без залучення мітохондрій.
Скупчення тригліцеридів в кардіоміоцитах пов'язана з зменшенням синтезу кардіоліпину і зміною дихальної функції мітохондрії, так як цитохром з окислотою комплексу IV ETS пов'язаний з кардіоліпином. Зміна структури мітохондріальної мембрани призводить до виходу цитохрому c і апоптозу без участі окислювачів. Таким чином, накопичення пальмової кислоти в клітинах нежирних тканин призводить до збільшення синтезу кераміду і зменшення синтезу кардіоліпину, що викликає апоптоз, і до зміни активності рецепторів.
У зв'язку з накопиченням тригліцеридів у клітинах (тригліцериди самі не викликають апоптозу) вважається спробою тіла, щоб уникнути ефекту ліпотоксичності.
Сфінгомієлін і пальмова кислота виявляє високу афінність для холестерину. Підвищення вмісту сфінгомієліну та пальмової кислоти в мембрані може пояснити вікове накопичення холестерину в мембранах, а також зміна чутливості рецептора інсуліну.
4961.31
Інсулінний рецептор асоціюється з мембранними рафтингами, а зміна складу рафтів впливає на його чутливість. Скупчення тригліцеридів у нежирних тканинах і пов'язаного зниження чутливості рецептора інсуліну призводить до виникнення інсулінорезистентності і гіперглікемії, тобто підвищеного вмісту глюкози в крові. Інсулін рецептор – кінази тирозин.
За допомогою автофосфориляції активуються різні доріжки, зокрема, PI-3-K шляхової дороги (фосінозитол-3-кенази), через які відбувається транспортування глюкози в клітинку, оскільки транспортер глюкози GLUT4 входить до його активного робочого стану.
У зв'язку з активним ліполізом вільних жирних кислот (FFA) і прозапальних цитокінів, вони впливають на субстрати рецептора інсуліну і тим самим блокують PI-3-K шляховий шлях, в результаті чого блокуються ефекти, що цей шлях має на метаболізмі глюкози, і глюкоза не може ввести клітинку. Таким чином, інсулінорезистентність розвивається, тобто надмірна кількість в'язкої жирової тканини блокує вхідний сигнал і призводить до того, що інсулінові рецептори стають нечутливими до інсуліну, а її біологічна роль спотворюється.
Залежно від індивідуальної чутливості та генетичності, інсулінорезистентність може розвиватися в різних тканинах. Надмірне ФФА повідомляє про прогресування інсулінорезистентності багатьох тканин - м'язів, включаючи міокард, гепатичний, адсипоз, а також ендотеліальні клітини, сприяє прогресуванню ішемічних змін в міокарда, включаючи зміни, пов'язані з порушенням бета-окислення ФФА в міокарда.
Крок п'ять: печінковий і м'язовий інсулінорезистентність.
В умовах підвищеного надходження їжі, багатих жирами і вуглеводами, стимулюється секреція інсуліну, яка, в свою чергу, активізує ліпогенез і відкладення ФФА в жировій тканині. Однак генетично визначається обмеження здатності накопичувати ліпіди, тому при об'ємі жирової тканини досягає максимального, надлишок ФФА починає входити в печінку і м'язи.
Надлишок ФФА супроводжується накопиченням тригліцеридів в пароенхімальних клітинах багатьох тканин, а саме в скелетних і кардіоміоцитах і в гепатоцитах, що призводить до їх пошкодження і хронічної дисфункції.
В результаті печінка в умовах інсулінорезистентності починає активно синтезувати жирні кислоти, тригліцериди, ліполіз прискорює, але вже в жировій тканині. Крім того, всі ті процеси виникають в печінці, які пригнічують пацієнта з вісцеральним ожирінням до цукрового діабету: глюкозогенез стимулюється і пригнічують гліколіз і синтез глікогену.

Досить складна схема виявлена, що відображає, що інсулінорезистентність, або інсулінорезистентність, призводить до того, що печінка перевантажується жирними кислотами.
Це пов'язано з тим, що активні жирні кислоти синтезуються в печінці, окислення жирних кислот знижується, жирні кислоти активно йдуть в печінку з вісцеральної жирової тканини і, крім того, жир, який надходить в нашу печінку в складі хіміомікронів, також перевантаження печінки з вільними жирними кислотами.
Ці процеси призводять до того, що печінка не здатна метаболізувати β-окислення ФФА, перекислення ліпідів відбувається, в результаті чого утворюються реактивні види кисню в великих кількостях, виникає окислювальний стрес, і це такі фактори, що призводять до фосфонії інсулінопроцедурної субстрату, які ми говорили про в попередньому слайді, тим самим, інсулінорезистентність знову викликається, тобто різновид замкненого кола, і вже важко визначити у пацієнта, що є первинним.
Крім того, доведено, що макрофаги в'язкої жирової тканини мають прозапальну активність. Крім того, CD-8 + Т-лімфоцити знаходяться в жировій тканині, яка активно секретує прозапальні цитокіни і, таким чином, в печінці в стані жирової дистрофії вже може переходити на наступний етап з розвитком NASH у хворих та інших наслідків, до яких ведеться NAFLD.

М'язова інсулінорезистентність.
Незабаром після печінки жир починає накопичуватися в м'язах. метаболічна патологія, характерна для ІР, є накопичення тригліцеридів навколо м'язових фібрилів. Однак скупчення тригліцеридів всередині скелетних м'язів не вважається безпосередньою причиною розвитку цукрового діабету 2, але, здається, є маркером ліпідних проміжних сполук, таких як ацетил КоА, кераміди і діакілгліцерин.
За останніми дослідженнями порушення шляхів передачі інсуліну в основному пов'язана з патологічним обміном ФФА в скелетних м'язових клітинах, які «не впоратися» з їх використанням при перевищенні ФФА. Дійсно, локальне накопичення всередині скелетних м'язів таких метаболітів ФФА як кераміди, діагліцерол або ацил-КоА призводить до порушення передавання інсулінових сигналів і, тим самим, до порушення глюкози транспорту.

Крок шість. Злий цикл інсулінорезистентності.
інсулінорезистентність, викликана високими рівнями ФФА, додатково збільшує концентрацію ФФА в плазмі. Як знайдено, інсулінорезистентні жирові клітини секрети підвищених рівнів ФФА. Це, фактично, дозволяє розглянути підвищені рівні ФФА як маркер ІР.
Дійсно, з ІР рівень ФФА в гепатоцитах зростає, тому що в них:
(1) підвищується ново-ліпогенез,
(2) естерифікація ФФА перевищує їх окислення,
(3) естерифіковані РК-дисплеї зберігаються як тригліцериди або відправлені для синтезу VLDL-X (багатих тригліцеридів),
При зменшенні інсулін-регульованої тригліцеридної мобілізації.
Інсуліностійкі асоціоцити інтенсивно порушують тригліцериди, що містяться в них і випускають ФФА, утворену з них в кровотік (як з ожирінням і без нього). Потік ФФА з жирових клітин підвищується і, крім того, ФФА також залишає X-VLDL і з плазмохімічних показників і через кровоплин частково спрямований на інші органи, а частково - назад до печінки, де вони знову перетворюються в тригліцериди. Є «пампінг» печінки ФФА і тригліцеридів. Це має найбільш серйозні наслідки.

Крок 7. Прискорений атеросклероз.
Підвищені рівні ФФА призводять до дисліпідемії та атерогенезу
Як інсулінорезистентність призводить до дисліпідемії Підвищені тригліцериди печінки стимулюють утворення Apo B і X-VLDL,
Ось як це відбувається.
1 час З печінки високі рівні VLDL-X виділяються в плазму, де ліполіз від VLDL-X утворює FFA і високоатеросогенний залишок (відмова) частинок тригліцерид-багатих ліпопротеїнів.
1 час Від плазми, FFA і залишків частинок знову поглинаються печінкою, яка додатково збільшує рівень ФФА в гепатоцитах і додатково стимулює синтез X-VLDL.
(1) У печінці з високим рівнем VLDL-X і нормальними рівнями білка CETP (холестериловий протеїн) - носієм холестерину ефіру, тригліцеридів від VLDL-X переходить в HDL-X, а холестерин від HDL-X переходить в VLDL-X. В результаті: (a) холестерин-багатий дуже атеросогенний залишок X-VLDL частинок і (b) HDL X, що містить багато тригліцеридів і невеликий холестерин.
(4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) (4) Такі частинки HDL-X втрачають тригліцериди (під впливом печінкової губи) і їх головна аполіпопротеїн Apo A1. В результаті знижується рівень антитерогенного X-HDL.
(5) (4) (5) (1) (5) (1) (5) (1) (5) (1) (5) (1) (5) (1) (5) (1) (5) (1) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (4) (5) (4) (5) (4) (5) (5) (4) (5) (4) (5) (5) (4) (5) (4) (5) (4) (5) (5) (4) (5) (4) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) (5) На високих рівнях VLDL-X (багаті тригліцериди), CETP несе тригліцериди від VLDL-X до LDL-X і холестерину від LDL-X до VLDL-X.
(6) Багаті тригліцериди X-LDL через активність печінкових або ліпопротеїнових ліпідів втрачають тригліцериди, зменшують розмір і стають дуже атеросогенними невеликими щільні частинки X-LDL.
Таким чином, підвищені рівні ФФА призводять до зменшення рівня «антиатерогенної» HDL-X, утворення вкрай атеросогенних дрібних, щільних LDL-C частинок і підвищення рівня тригліцериду плазми.

Але є ще один спосіб, що високий рівень FFA викликає атерогенез. Цей шлях є більш прямим і коротким. Підвищений рівень ФФА в ІР викликає в мітохондрії макросудинних ендотеліальних клітин надсинтез реактивних видів кисню, що призводить до окислення ЛЛ-Х і модифікації HDL-X. Це викликає запальний процес в стінках судин, утворення і накопичення холестеринових нальотів і, в результаті ішемії. Видання
Джерела.
1. Публікації В.В. Велкова
2. В.В. Велков. Діагностика праці. - 2009. - Т. 3, No 49. - С. 55-71.
3. У Вельков В.В. Вільні жирні кислоти – фактор ризику інсулінорезистентності та ішемії: перспективи оцінки ризику та діагностики
4. Л.І. Данилова феномен інсулінорезистентності в клінічній практиці: механізми формування та можливості корекції
5. Умань Е.В. Терешина Роль жирних кислот у розвитку вікового окислювального стресу. Пороги в геронології, 2007. Т. 20, No 1
6. Ендокринологія та метаболізм, Ф. Феліг, Д. Бакстер
Автор: Андрій Білловкін
Психосоматика захворювань шиї: Через наші горлі ми ковтати реальність.
Що не варто лікувати
Джерело: www.beloveshkin.com/2016/01/svobodnye-zhirnye-kisloty-klyuch-k-ponimaniyu-insulinorezistentnosti.html