475
0.1
2016-09-20
Метаболическая память: месть подается холодной
Подпишитесь на наш канал в Телеграм чтобы не пропустить интересную статью!
Гликация (гликирование) приводит не только к структурным, но и функциональными изменениям многих элементов организма. Более того, эти изменения оказываются долгосрочными, от нескольких месяцев до нескольких лет (6 лет и более). Этот феномен получил название «метаболическая память», сегодня я расскажу, как гликация может перестроить нормальный обмен клеток и почему изменение образа жизни может не всегда сработать сразу эффективно.
Гликация догоживущих белков и ДНК — это процесс, незаметный вначале, но очень трудноисправимый потом. Да, конечные продукты гликозилирования очень мстительны, и месть — это блюдо, которое подается холодным.

Что такое метаболическая память?
Термин «метаболическая память» появился при изучении сахарного диабета. Дело в том, что, несмотря на достигнутые успехи в лечении заболевания – разработку новых сахароснижающих средств, достижение успешного контроля гликемии и определение маркеров уровня гликемии, у большинства пациентов развиваются серьезные поражения органов-мишеней, приводящие к тяжелым осложнениям.
Метаболическая память (менее известная как гипергликемическая или сосудистая память) – это эффект, приводящий к развитию долгосрочных последствий длительного периода плохо компенсированного уровня глюкозы крови. Другими словами, чем дольше диабет остается некомпенсированным, тем дольше сохраняется этот разрушающий эффект, даже после улучшения контроля над заболеванием.
Начальные, ранние эпизоды гипергликемии оказывают негативное воздействие на уровне клеток сетчатки, почек, а также на функционирование нервных волокон. «Метаболическая память» — это такая ситуация, когда повышение гликемии — ещё до диагностирования заболевания — уже патологически сказывается на различных системах организма, в первую очередь, на нервных волокнах. Более того, если гликемический контроль долгое время был недостаточным, повреждающее действие может сохраняться и в будущем, даже после оптимизации уровня глюкозы.
В частности, в обширном исследовании ADVANCE было выявлено, что строгий контроль гликемии при сахарном диабете (СД) 2 типа приводил только к уменьшению частоты возникновения нефропатии, при этом частота развития ретинопатии и макрососудистых осложнений не снижалась. Результаты другого масштабного исследования ACCORD продемонстрировали, что строгий гликемический контроль привел к росту летальности среди пациентов, страдающих СД 2 типа.
В исследовании UKPDS не было выявлено достоверного влияния жесткого гликемического контроля на инфаркт миокарда. Перспективным направлением является подход, основанный на предположении, что гипергликемия оказывает долговременный повреждающий эффект при сахарном диабете как 1, так и 2 типов и что строгий гликемический контроль, если он не обеспечивается на очень ранних стадиях заболевания, не является достаточным для полного предотвращения осложнений.
В основе данной гипотезы лежит представление о феномене метаболической памяти, которое заключается в том, что ранний гликемический фон «запоминается» в тканях и органах-мишенях (сетчатка глаз, почки, сердце, периферические нервы). Первые данные, подтвердившие существование метаболической памяти, были получены в опытах на животных при изучении клеток сетчатки собак, одна группа которых была переведена на строгий контроль гликемии после двухмесячного, а другая – после 2,5-летнего периода гипергликемии.
Исследование сетчатки проводилось через 5 лет после начала эксперимента. Парадоксально, что у животных, у которых контроль гликемии начался через 2 месяца, наблюдались незначительные признаки ретинопатии, так же как и у животных с контролем гликемии на протяжении всего периода исследования. В то же время у собак, которые были переведены на строгий гликемический контроль через 2,5 года, частота ретинопатии была сходной с уровнем ретинопатии собак, находившихся в состоянии гипергликемии все 5 лет.
Термин «метаболическая память» имеет несколько синонимов – гликемическая память, гипергликемическая память, наследственный эффект и др… Клиническое подтверждение феномена метаболической памяти было получено в ходе масштабного клинического исследования СД 1 типа DCCT (Diabetes Complications and Control Trial) и последующего исследования EDIC (Epidemiology of Diabetes Interventions and Complications). В ходе DCCT пациенты с сахарным диабетом 1 типа были разделены на 2 группы – интенсифицированной инсуинотерапии и традиционной терапии, направленной на нормализацию гликемии.
В дальнейшем в ходе исследования EDIC, проводимого на той же популяции пациентов, было выявлено, что среди участников, которым проводилась стандартная терапия в ходе DCCT, распространенность микрососудистых осложнений, таких как нефропатия и ретинопатия, была выше по сравнению с пациентами группы интенсифицированной терапии, хотя через 6,5 лет все участники исследования были переведены на интенсифицированную терапию.
Другой интересный факт – в обеих группах средний уровень гликированного гемоглобина был практически эквивалентным. Кроме того, недавние результаты EDIC подтверждают долговременное влияние раннего гликемического контроля на прогрессирование макрососудистых осложнений и сердечно-сосудистых заболеваний.
Феномен метаболической памяти также был выявлен при сахарном диабете 2 типа по результатам исследования UKPDS (U.K. Prospective Diabetes Study). У пациентов с СД 2 типа, которые находились на стандартной терапии в течение исследования, уровень микрососудистых и сердечно-сосудистых осложнений был выше по сравнению с больными, получавшими интенсивную терапию.
Повреждающее влияние гипергликемии на клеточном уровне реализуется с помощью четырех хорошо известных механизмов: индукция полиолового пути, реализуемого через фермент альдозоредуктазу; увеличение формирования конечных продуктов избыточного гликозилирования (AGE); активация протеинкиназы С; активация гексозаминового пути.
Значимость каждого из перечисленных механизмов была неоднократно подтверждена в ходе исследований на животных с использованием различных ингибиторов того или иного пути биохимических реакций, что позволило предотвратить диабетические осложнения. Однако проблема заключалась в том, что, несмотря на очевидные успехи применения ингибиторов в экспериментах на животных, при использовании тех же препаратов в лечении пациентов результаты были крайне неудовлетворительными.
В ходе дальнейших исследований было установлено, что все четыре патологические реакции запускаются в ответ на повышенную продукцию супероксид-аниона в дыхательной цепи митохондрий – процесс, индуцированный гипергликемией. Образование супероксида происходит под влиянием повышенной внутриклеточной концентрации глюкозы, нарушения работы компонентов дыхательной цепи митохондрий и переноса электронов на молекулу кислорода.
Если генерация большого количества супероксид-аниона – это ключевой процесс в развитии диабетических осложнений, то насколько важную роль он играет в феномене метаболической памяти? Супероксид, как и большинство реактивных молекул (радикалов), обладает очень коротким периодом полураспада – не дольше минуты, тогда как гликемическая память может длиться годами.
На самом деле мишенями для супероксид-ионов и других радикалов служат нуклеиновые кислоты, белки, липиды и липопротеиды с долгим периодом полураспада. Эти молекулы, поврежденные радикалами, способны нарушать работу клеток в течение долгого времени.
Соединение 3-нитрозин, продукт взаимодействия свободных радикалов с клеточными протеинами, может служить важным маркером окислительного стресса при осложнениях диабета. Данное вещество было использовано в многочисленных экспериментах на животных моделях, где была продемонстрирована роль оксидативного стресса в формировании гликемической памяти.
Например, R.A. Kowluru и соавт. установили, что у крыс со стрептозотоцин-индуцированным диабетом нормализация уровня гликемии в течение 6 месяцев вслед за 6-месячным периодом гипергликемии не приводила к достоверному снижению уровня 3-нитрозина в клетках сетчатки, так же как и у животных, у которых гипергликемия поддерживалась на протяжении всего исследования. Еще раз обращаю ваше внимание – за полгода ничего не изменилось, и это у крысы!
АGE-подукты – это субстрат метаболической памяти.
Наряду с оксидативным стрессом митохондрии клеток подвержены другому негативному воздействию гипергликемии – гликированию митохондриальных протеинов. В частности, установлено, что уровень метилглиоксаля – высокореактивного побочного продукта гликолиза – оказывается повышенным у пациентов, страдающих диабетом.
Метилглиоксаль, реагируя с белками и нуклеиновыми кислотами клетки, образует так называемые конечные продукты избыточного гликозилирования – advanced glycation end-products (AGE). AGE-продукты играют ключевую роль в развитии диабетических осложнений.
Гликирование белков дыхательной цепи митохондрий приводит к нарушению ее работы и поддержанию образования избытка супероксид-ионов независимо от уровня гликемии. Кроме того, формирование AGE-продуктов в структуре митохондрий, будучи необратимым процессом, также может объяснять длительное существование метаболической памяти.
Показано, что у больных диабетом уровень AGE-продуктов в тканях повышен по сравнению со здоровыми людьми. Причем у пациентов с диабетом без осложнений уровень AGE-продуктов повышен на 20–30%, а при диабете в сочетании с сердечно-сосудистыми осложнениями или микроальбуминурией – на 40–100% по сравнению со здоровыми людьми.
В ходе исследования EDIC уровень AGE-продуктов, определяемый путем биопсии кожи, достоверно коррелировал с распространенностью ретинопатии и нефропатии. В отличие от гликированного гемоглобина, который при снижении гликемии подвергается частичному энзиматическому дегликозилированию, уровень других AGE-продуктов не зависит от текущего уровня глюкозы.
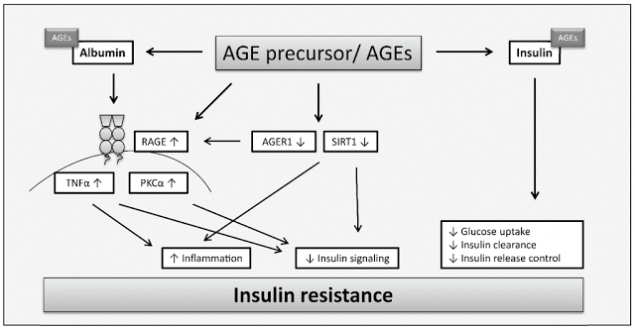
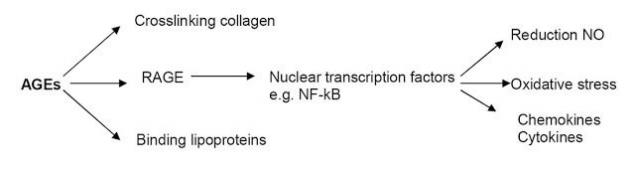
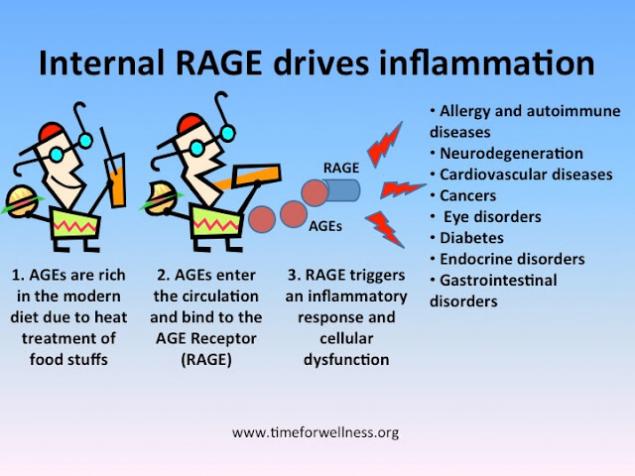
Кроме непосредственного участия AGE-продуктов, в формировании метаболической памяти играют роль их рецепторы, в частности тип рецепторов, известный под названием RAGE, который относится к суперсемейству иммуноглобулинов поверхностных клеточных молекул.
Связывание AGE и данного рецептора приводит к образованию активных форм кислорода (АФК) с последующей активацией чувствительного к окислению фактора транскрипции NF-κB в сосудистой стенке, регулирующего экспрессию воспалительных и «отвечающих на повреждение» генов, и непосредственно гена RAGE.
Данные события приводят в конечном итоге к эндотелиальной дисфункции и, как следствие, к вазоконстрикции, воспалительным явлениям, утолщению базальной мембраны и снижению способности к вазодилатации. Негативное действие AGE-продуктов реализуется не только в стенках сосудах, но и в нейронах и даже в костной ткани. В нейронах AGE-продукты провоцируют постепенное разрушение нервных волокон. Гликирование белков дыхательной цепи митохондрий в совокупности с повреждением митохондриальной ДНК могут приводить к образованию новых генераций АФК, персистирующему оксидативному стрессу и клеточному повреждению.
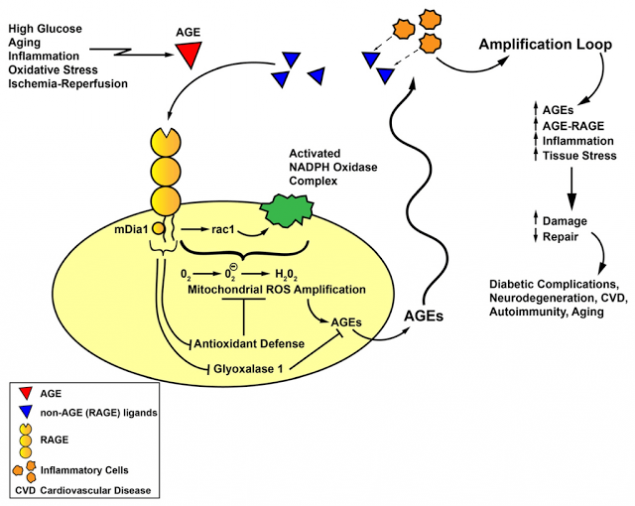
Порочный круг: АGE-RAGE-воспаление-стресс.
Данный самоподдерживающийся процесс лежит в основе метаболической памяти – ведущего механизма патогенеза диабетических осложнений, который не зависит от текущего уровня гликемии. В этой связи весьма перспективными в плане предотвращения развития осложнений представляются методы «выключения» метаболической памяти.
Ранее большие надежды возлагались на применение антиоксидантов, поскольку ключевую роль в механизме повреждения играет оксидативный стресс, однако клиническое применение данных препаратов при диабете не привело к значимым улучшениям. Тем не менее предполагается, что подавление образования AGE-продуктов, экспрессии RAGE и оксидативного стресса в сочетании с нормализацией уровня гликемии эффективно для предотвращения осложнений.
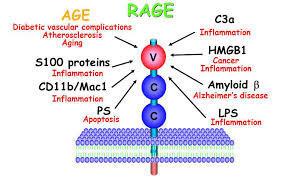
AGE также могут нарушить функции клеток, связывая собой различные рецепторы, включая макрофаги, клетки эндотелия и гладких мышц, клетки в тканях почек, а также нервные клетки. Такие рецепторы, вступающие во взаимодействие с конечными продуктами гликации, называются RAGE-рецепторами. RAGE- рецепторы являются ключевыми медиаторами в реакции тела на бактерии и вирусы.
AGE, гликозилированные белки, липиды и даже нуклеиновые кислоты могут выступать в качестве лигандов, взаимодействующих с RAGE-рецепторами и инициирующих внутриклеточные сигналы: например, активацию NF-kB – основного воспалительного агента, способствующего старению.
Вместо того, чтобы присоединяться напрямую к протеинам типа коллагена или эластина, эти растворимые AGE выступают в роли лигандов на RAGE-рецепторах и провоцируют воспалительный процесс. RAGE-рецепторы поддерживают клетку в активированном состоянии, из-за чего кратковременные противовоспалительные реакции переходят в продолжительную клеточную дисфункцию.
АGE-подукты и рецептор к ним (RAGE): от сосудов до Альцгеймера.
Рецептор конечных продуктов гликозилирования (RAGE) является мультилигандом трансмембранного гликопротеина типа I, принадлежащего суперсемейству иммуноглобулина (Ig). RAGE может участвовать в ряде патологических процессов, включая диабет, болезнь Альцгеймера (БА), системный амилоидоз и опухолевый рост.
RAGE изначально охарактеризован на основе его способности связывать AGEs, аддукты, образованные неферментативным гликозилированием и окислением белков и липидов. Этот процесс происходит при нормальном процессе старения и резко ускоряется при диабете, где гипергликемия служит основным триггером. Существенные доказательства подтверждают роль взаимодействий AGE/RAGE в патофизиологии диабета. Оба: и AGEs, и RAGE повышены при диабете в кровеносных сосудах, моноцитах и подоцитах.
RAGE может также опосредовать физиологические функции, такие как рост нейронов, выживание и регенерацию, и играет важную роль в провоспалительных реакциях. RAGE экспрессируется на высоком уровне в процессе развития, особенно в центральной нервной системе (ЦНС). Он также экспрессируется на более низком уровне в клетках взрослого организма: клетках эндотелия, в том числе и гладкомышечных клетках, в мононуклеарных фагоцитах, перицитах, нейронах, кардиомиоцитах, гепатоцитах. RAGE лиганды включают конечные продукты гликозилирования (AGEs), белок амилоида-β (Aβ), HMG-1 (также известный как амфотерин) и несколько членов суперсемейства протеина S100.
Блокада активации RAGE рекомбинантным sRAGE или блокирующими функцию антителами снижает сосудистую проницаемость, развитие атеросклеротических поражений и усиливает заживление ран у больных сахарным диабетом грызунов. У RAGE-нулевых мышей с диабетом не развиваются признаки нефропатии, включая усиленную экспансию мезангиального матрикса и утолщение гломерулярной мембраны.
И, напротив, трансгенная гиперэкспрессия RAGE приводит к обострению диабетической нефропатии и ретинопатии. RAGE является также рецептором для β-складчатых фибрилл, структур, которые характерны для амилоида. Aβ является основным компонентом нейродегенеративных бляшек, связанных с болезнью Альцгеймера (БА), и RAGE колокализуется с Aβ в мозге больных БА в повышенных количествах.
С молекулярной точки зрения болезнь Альцгеймера характеризуется отложениями аномально агрегированных белков. В случае внеклеточных амилоидных бляшек такие отложения главным образом состоят из филаментов β-амилоидных пептидов (Aβ), и в случае внутриклеточных нейрофибриллярных клубков (NFT), главным образом, из белка tau.
AD также характеризуется повышенной экспрессией RAGE в нейронах. RAGE является рецептором семейства иммуноглобулинов, имеющим много лигандов, который функционирует в качестве передающего сигнал акцептора Aβ на клеточной поверхности. Рецептор конечных продуктов гликозилирования (RAGE), который локализован не только на нейронах, астроглии и микроглии, но и на эндотелиальных клетках, где выполняет роль транспортера бета-амилоида в мозг из кровяного русла.
Несколькими группами показано, что инфузия Aβ40 у мышей приводит к вазоконстрикции сосудов головного мозга и ослаблению мозгового кровообращения (CBF). Пациенты, страдающие AD, также имеют ослабленное мозговое кровообращение. В мышиных моделях AD, в которых трансгенные животные сверхэкспрессируют белок амилоидного предшественника (APP), который приводит к заболеванию, вызывая образование бляшек, показано, что RAGE вовлечен в качестве патогенного фактора в прогрессирование заболевания.
Показано, что RAGE связывается с Aβ-пептидами. Ингибирование такого взаимодействия подавляет накопление Aβ у трансгенных животных в модели; поэтому полагают, что RAGE вовлечен в AD. Показано, что лечение с использованием sRAGE (растворимого RAGE), а также анти-RAGE-антителами снижает количество бляшек (Deane et al, 2003). Блокирование взаимодействия RAGE с амилоидом с помощью антител может быть подходящим способом лечения пациентов с AD; однако, существующие поликлональные антитела, полученные из сыворотки животных, не подходят для хронического лечения человека.опубликовано
Автор: Андрей Беловешкин
Читайте также:
Антипаразитарные чистки и затравки
Можно лечить нервную систему и гипертонию, а получить инсульт из-за остехондроза!
P.S. И помните, всего лишь изменяя свое потребление — мы вместе изменяем мир! ©
Присоединяйтесь к нам в Facebook , ВКонтакте, Одноклассниках
Источник: www.beloveshkin.com/2016/05/glikirovanie-i-metabolicheskaya-pamyat-mest-podaetsya-kholodnoj.html
Гликация догоживущих белков и ДНК — это процесс, незаметный вначале, но очень трудноисправимый потом. Да, конечные продукты гликозилирования очень мстительны, и месть — это блюдо, которое подается холодным.

Что такое метаболическая память?
Термин «метаболическая память» появился при изучении сахарного диабета. Дело в том, что, несмотря на достигнутые успехи в лечении заболевания – разработку новых сахароснижающих средств, достижение успешного контроля гликемии и определение маркеров уровня гликемии, у большинства пациентов развиваются серьезные поражения органов-мишеней, приводящие к тяжелым осложнениям.
Метаболическая память (менее известная как гипергликемическая или сосудистая память) – это эффект, приводящий к развитию долгосрочных последствий длительного периода плохо компенсированного уровня глюкозы крови. Другими словами, чем дольше диабет остается некомпенсированным, тем дольше сохраняется этот разрушающий эффект, даже после улучшения контроля над заболеванием.
Начальные, ранние эпизоды гипергликемии оказывают негативное воздействие на уровне клеток сетчатки, почек, а также на функционирование нервных волокон. «Метаболическая память» — это такая ситуация, когда повышение гликемии — ещё до диагностирования заболевания — уже патологически сказывается на различных системах организма, в первую очередь, на нервных волокнах. Более того, если гликемический контроль долгое время был недостаточным, повреждающее действие может сохраняться и в будущем, даже после оптимизации уровня глюкозы.
В частности, в обширном исследовании ADVANCE было выявлено, что строгий контроль гликемии при сахарном диабете (СД) 2 типа приводил только к уменьшению частоты возникновения нефропатии, при этом частота развития ретинопатии и макрососудистых осложнений не снижалась. Результаты другого масштабного исследования ACCORD продемонстрировали, что строгий гликемический контроль привел к росту летальности среди пациентов, страдающих СД 2 типа.
В исследовании UKPDS не было выявлено достоверного влияния жесткого гликемического контроля на инфаркт миокарда. Перспективным направлением является подход, основанный на предположении, что гипергликемия оказывает долговременный повреждающий эффект при сахарном диабете как 1, так и 2 типов и что строгий гликемический контроль, если он не обеспечивается на очень ранних стадиях заболевания, не является достаточным для полного предотвращения осложнений.

В основе данной гипотезы лежит представление о феномене метаболической памяти, которое заключается в том, что ранний гликемический фон «запоминается» в тканях и органах-мишенях (сетчатка глаз, почки, сердце, периферические нервы). Первые данные, подтвердившие существование метаболической памяти, были получены в опытах на животных при изучении клеток сетчатки собак, одна группа которых была переведена на строгий контроль гликемии после двухмесячного, а другая – после 2,5-летнего периода гипергликемии.
Исследование сетчатки проводилось через 5 лет после начала эксперимента. Парадоксально, что у животных, у которых контроль гликемии начался через 2 месяца, наблюдались незначительные признаки ретинопатии, так же как и у животных с контролем гликемии на протяжении всего периода исследования. В то же время у собак, которые были переведены на строгий гликемический контроль через 2,5 года, частота ретинопатии была сходной с уровнем ретинопатии собак, находившихся в состоянии гипергликемии все 5 лет.
Термин «метаболическая память» имеет несколько синонимов – гликемическая память, гипергликемическая память, наследственный эффект и др… Клиническое подтверждение феномена метаболической памяти было получено в ходе масштабного клинического исследования СД 1 типа DCCT (Diabetes Complications and Control Trial) и последующего исследования EDIC (Epidemiology of Diabetes Interventions and Complications). В ходе DCCT пациенты с сахарным диабетом 1 типа были разделены на 2 группы – интенсифицированной инсуинотерапии и традиционной терапии, направленной на нормализацию гликемии.
В дальнейшем в ходе исследования EDIC, проводимого на той же популяции пациентов, было выявлено, что среди участников, которым проводилась стандартная терапия в ходе DCCT, распространенность микрососудистых осложнений, таких как нефропатия и ретинопатия, была выше по сравнению с пациентами группы интенсифицированной терапии, хотя через 6,5 лет все участники исследования были переведены на интенсифицированную терапию.
Другой интересный факт – в обеих группах средний уровень гликированного гемоглобина был практически эквивалентным. Кроме того, недавние результаты EDIC подтверждают долговременное влияние раннего гликемического контроля на прогрессирование макрососудистых осложнений и сердечно-сосудистых заболеваний.
Феномен метаболической памяти также был выявлен при сахарном диабете 2 типа по результатам исследования UKPDS (U.K. Prospective Diabetes Study). У пациентов с СД 2 типа, которые находились на стандартной терапии в течение исследования, уровень микрососудистых и сердечно-сосудистых осложнений был выше по сравнению с больными, получавшими интенсивную терапию.
Повреждающее влияние гипергликемии на клеточном уровне реализуется с помощью четырех хорошо известных механизмов: индукция полиолового пути, реализуемого через фермент альдозоредуктазу; увеличение формирования конечных продуктов избыточного гликозилирования (AGE); активация протеинкиназы С; активация гексозаминового пути.
Значимость каждого из перечисленных механизмов была неоднократно подтверждена в ходе исследований на животных с использованием различных ингибиторов того или иного пути биохимических реакций, что позволило предотвратить диабетические осложнения. Однако проблема заключалась в том, что, несмотря на очевидные успехи применения ингибиторов в экспериментах на животных, при использовании тех же препаратов в лечении пациентов результаты были крайне неудовлетворительными.
В ходе дальнейших исследований было установлено, что все четыре патологические реакции запускаются в ответ на повышенную продукцию супероксид-аниона в дыхательной цепи митохондрий – процесс, индуцированный гипергликемией. Образование супероксида происходит под влиянием повышенной внутриклеточной концентрации глюкозы, нарушения работы компонентов дыхательной цепи митохондрий и переноса электронов на молекулу кислорода.
Если генерация большого количества супероксид-аниона – это ключевой процесс в развитии диабетических осложнений, то насколько важную роль он играет в феномене метаболической памяти? Супероксид, как и большинство реактивных молекул (радикалов), обладает очень коротким периодом полураспада – не дольше минуты, тогда как гликемическая память может длиться годами.
На самом деле мишенями для супероксид-ионов и других радикалов служат нуклеиновые кислоты, белки, липиды и липопротеиды с долгим периодом полураспада. Эти молекулы, поврежденные радикалами, способны нарушать работу клеток в течение долгого времени.
Соединение 3-нитрозин, продукт взаимодействия свободных радикалов с клеточными протеинами, может служить важным маркером окислительного стресса при осложнениях диабета. Данное вещество было использовано в многочисленных экспериментах на животных моделях, где была продемонстрирована роль оксидативного стресса в формировании гликемической памяти.
Например, R.A. Kowluru и соавт. установили, что у крыс со стрептозотоцин-индуцированным диабетом нормализация уровня гликемии в течение 6 месяцев вслед за 6-месячным периодом гипергликемии не приводила к достоверному снижению уровня 3-нитрозина в клетках сетчатки, так же как и у животных, у которых гипергликемия поддерживалась на протяжении всего исследования. Еще раз обращаю ваше внимание – за полгода ничего не изменилось, и это у крысы!
АGE-подукты – это субстрат метаболической памяти.
Наряду с оксидативным стрессом митохондрии клеток подвержены другому негативному воздействию гипергликемии – гликированию митохондриальных протеинов. В частности, установлено, что уровень метилглиоксаля – высокореактивного побочного продукта гликолиза – оказывается повышенным у пациентов, страдающих диабетом.
Метилглиоксаль, реагируя с белками и нуклеиновыми кислотами клетки, образует так называемые конечные продукты избыточного гликозилирования – advanced glycation end-products (AGE). AGE-продукты играют ключевую роль в развитии диабетических осложнений.
Гликирование белков дыхательной цепи митохондрий приводит к нарушению ее работы и поддержанию образования избытка супероксид-ионов независимо от уровня гликемии. Кроме того, формирование AGE-продуктов в структуре митохондрий, будучи необратимым процессом, также может объяснять длительное существование метаболической памяти.

Показано, что у больных диабетом уровень AGE-продуктов в тканях повышен по сравнению со здоровыми людьми. Причем у пациентов с диабетом без осложнений уровень AGE-продуктов повышен на 20–30%, а при диабете в сочетании с сердечно-сосудистыми осложнениями или микроальбуминурией – на 40–100% по сравнению со здоровыми людьми.
В ходе исследования EDIC уровень AGE-продуктов, определяемый путем биопсии кожи, достоверно коррелировал с распространенностью ретинопатии и нефропатии. В отличие от гликированного гемоглобина, который при снижении гликемии подвергается частичному энзиматическому дегликозилированию, уровень других AGE-продуктов не зависит от текущего уровня глюкозы.
Кроме непосредственного участия AGE-продуктов, в формировании метаболической памяти играют роль их рецепторы, в частности тип рецепторов, известный под названием RAGE, который относится к суперсемейству иммуноглобулинов поверхностных клеточных молекул.
Связывание AGE и данного рецептора приводит к образованию активных форм кислорода (АФК) с последующей активацией чувствительного к окислению фактора транскрипции NF-κB в сосудистой стенке, регулирующего экспрессию воспалительных и «отвечающих на повреждение» генов, и непосредственно гена RAGE.
Данные события приводят в конечном итоге к эндотелиальной дисфункции и, как следствие, к вазоконстрикции, воспалительным явлениям, утолщению базальной мембраны и снижению способности к вазодилатации. Негативное действие AGE-продуктов реализуется не только в стенках сосудах, но и в нейронах и даже в костной ткани. В нейронах AGE-продукты провоцируют постепенное разрушение нервных волокон. Гликирование белков дыхательной цепи митохондрий в совокупности с повреждением митохондриальной ДНК могут приводить к образованию новых генераций АФК, персистирующему оксидативному стрессу и клеточному повреждению.

Порочный круг: АGE-RAGE-воспаление-стресс.
Данный самоподдерживающийся процесс лежит в основе метаболической памяти – ведущего механизма патогенеза диабетических осложнений, который не зависит от текущего уровня гликемии. В этой связи весьма перспективными в плане предотвращения развития осложнений представляются методы «выключения» метаболической памяти.
Ранее большие надежды возлагались на применение антиоксидантов, поскольку ключевую роль в механизме повреждения играет оксидативный стресс, однако клиническое применение данных препаратов при диабете не привело к значимым улучшениям. Тем не менее предполагается, что подавление образования AGE-продуктов, экспрессии RAGE и оксидативного стресса в сочетании с нормализацией уровня гликемии эффективно для предотвращения осложнений.

AGE также могут нарушить функции клеток, связывая собой различные рецепторы, включая макрофаги, клетки эндотелия и гладких мышц, клетки в тканях почек, а также нервные клетки. Такие рецепторы, вступающие во взаимодействие с конечными продуктами гликации, называются RAGE-рецепторами. RAGE- рецепторы являются ключевыми медиаторами в реакции тела на бактерии и вирусы.
AGE, гликозилированные белки, липиды и даже нуклеиновые кислоты могут выступать в качестве лигандов, взаимодействующих с RAGE-рецепторами и инициирующих внутриклеточные сигналы: например, активацию NF-kB – основного воспалительного агента, способствующего старению.
Вместо того, чтобы присоединяться напрямую к протеинам типа коллагена или эластина, эти растворимые AGE выступают в роли лигандов на RAGE-рецепторах и провоцируют воспалительный процесс. RAGE-рецепторы поддерживают клетку в активированном состоянии, из-за чего кратковременные противовоспалительные реакции переходят в продолжительную клеточную дисфункцию.
АGE-подукты и рецептор к ним (RAGE): от сосудов до Альцгеймера.
Рецептор конечных продуктов гликозилирования (RAGE) является мультилигандом трансмембранного гликопротеина типа I, принадлежащего суперсемейству иммуноглобулина (Ig). RAGE может участвовать в ряде патологических процессов, включая диабет, болезнь Альцгеймера (БА), системный амилоидоз и опухолевый рост.
RAGE изначально охарактеризован на основе его способности связывать AGEs, аддукты, образованные неферментативным гликозилированием и окислением белков и липидов. Этот процесс происходит при нормальном процессе старения и резко ускоряется при диабете, где гипергликемия служит основным триггером. Существенные доказательства подтверждают роль взаимодействий AGE/RAGE в патофизиологии диабета. Оба: и AGEs, и RAGE повышены при диабете в кровеносных сосудах, моноцитах и подоцитах.
RAGE может также опосредовать физиологические функции, такие как рост нейронов, выживание и регенерацию, и играет важную роль в провоспалительных реакциях. RAGE экспрессируется на высоком уровне в процессе развития, особенно в центральной нервной системе (ЦНС). Он также экспрессируется на более низком уровне в клетках взрослого организма: клетках эндотелия, в том числе и гладкомышечных клетках, в мононуклеарных фагоцитах, перицитах, нейронах, кардиомиоцитах, гепатоцитах. RAGE лиганды включают конечные продукты гликозилирования (AGEs), белок амилоида-β (Aβ), HMG-1 (также известный как амфотерин) и несколько членов суперсемейства протеина S100.
Блокада активации RAGE рекомбинантным sRAGE или блокирующими функцию антителами снижает сосудистую проницаемость, развитие атеросклеротических поражений и усиливает заживление ран у больных сахарным диабетом грызунов. У RAGE-нулевых мышей с диабетом не развиваются признаки нефропатии, включая усиленную экспансию мезангиального матрикса и утолщение гломерулярной мембраны.
И, напротив, трансгенная гиперэкспрессия RAGE приводит к обострению диабетической нефропатии и ретинопатии. RAGE является также рецептором для β-складчатых фибрилл, структур, которые характерны для амилоида. Aβ является основным компонентом нейродегенеративных бляшек, связанных с болезнью Альцгеймера (БА), и RAGE колокализуется с Aβ в мозге больных БА в повышенных количествах.

С молекулярной точки зрения болезнь Альцгеймера характеризуется отложениями аномально агрегированных белков. В случае внеклеточных амилоидных бляшек такие отложения главным образом состоят из филаментов β-амилоидных пептидов (Aβ), и в случае внутриклеточных нейрофибриллярных клубков (NFT), главным образом, из белка tau.
AD также характеризуется повышенной экспрессией RAGE в нейронах. RAGE является рецептором семейства иммуноглобулинов, имеющим много лигандов, который функционирует в качестве передающего сигнал акцептора Aβ на клеточной поверхности. Рецептор конечных продуктов гликозилирования (RAGE), который локализован не только на нейронах, астроглии и микроглии, но и на эндотелиальных клетках, где выполняет роль транспортера бета-амилоида в мозг из кровяного русла.
Несколькими группами показано, что инфузия Aβ40 у мышей приводит к вазоконстрикции сосудов головного мозга и ослаблению мозгового кровообращения (CBF). Пациенты, страдающие AD, также имеют ослабленное мозговое кровообращение. В мышиных моделях AD, в которых трансгенные животные сверхэкспрессируют белок амилоидного предшественника (APP), который приводит к заболеванию, вызывая образование бляшек, показано, что RAGE вовлечен в качестве патогенного фактора в прогрессирование заболевания.
Показано, что RAGE связывается с Aβ-пептидами. Ингибирование такого взаимодействия подавляет накопление Aβ у трансгенных животных в модели; поэтому полагают, что RAGE вовлечен в AD. Показано, что лечение с использованием sRAGE (растворимого RAGE), а также анти-RAGE-антителами снижает количество бляшек (Deane et al, 2003). Блокирование взаимодействия RAGE с амилоидом с помощью антител может быть подходящим способом лечения пациентов с AD; однако, существующие поликлональные антитела, полученные из сыворотки животных, не подходят для хронического лечения человека.опубликовано
Автор: Андрей Беловешкин
Читайте также:
Антипаразитарные чистки и затравки
Можно лечить нервную систему и гипертонию, а получить инсульт из-за остехондроза!
P.S. И помните, всего лишь изменяя свое потребление — мы вместе изменяем мир! ©
Присоединяйтесь к нам в Facebook , ВКонтакте, Одноклассниках
Источник: www.beloveshkin.com/2016/05/glikirovanie-i-metabolicheskaya-pamyat-mest-podaetsya-kholodnoj.html
Bashny.Net. Перепечатка возможна при указании активной ссылки на данную страницу.