886
5 most unimaginable human genetic pathologies
Sometimes the same is ... .
fatal familial insomnia, children's age and the extra bone in the wrong places - it's not only the ideas, inspirational filmmakers, but also real hereditary diseases that occur as a result of genetic mutations. "Theory and Practice" compiled a list of the most unimaginable diseases and describe their causes.
Fatal familial bessonnitsa
Fatal familial insomnia - a rare hereditary disease in which a person dies from an inability to sleep. Until now, it was observed only forty families around the world. Fatal Insomnia usually manifests between 30 and 60 years (usually - after 50 years) and extends from 7 to 36 months. As the disease progresses, the patient suffers from a more serious sleep disorders, and no sleeping pills do not help him. The first stage is accompanied by insomnia, panic attacks and phobias, the second added to them hallucinations and increased sweating. At the third stage of the disease the person loses the ability to sleep and begins to look much older than his years. Then developing dementia, and the patient dies - usually from starvation or pneumonia.
Lethal insomnia arises from the fact that in a codon (encoding trinucleotide) 178 gene PRNP, located at 20 chromosome instead of asparagine, which is normally present in the body structure of proteins and is required for normal functioning of the nervous system, appears aspartic acid. This changes the shape of the protein molecule, and it turns into a prion - aggressive abnormal protein in the composition which have nucleic acids. Under the influence of the surrounding prion molecules become like him, and this leads to irreversible changes. Before that, they occur in the tissues of the thalamus: subcortical "stations" of all kinds of sensitivity, including responsible for the occurrence of somnolence and motor functions: swallowing, sucking, chewing, laughing. Under the action of prions thalamic nuclei covered pores turn into a sponge and stop working.
The disease is characterized by an autosomal dominant mode of inheritance: that is, it does not have media. To children it is transmitted from parents with a probability of 50%, and only on condition that one of them is sick. Men and women suffer from fatal familial insomnia with the same frequency. Today the disease is considered incurable.
Narcolepsy-katapleksiya
Narcolepsy-cataplexy syndrome, which is characterized by sudden onset of sleep and relaxation of the muscles of the body, too, has a genetic nature and arises from the rapid phase of sleep disorders. It occurs more often fatal familial insomnia: at forty out of every one hundred thousand people, equally men and women. A person suffering from narcolepsy, suddenly able to sleep for a few minutes in the middle of the day. "Sleepy attacks" remind REM sleep and can happen very often - up to a hundred times a day, preceded by a headache or without. They are often triggered by inactivity, but can occur at exactly the wrong time during intercourse, sports, driving. Wakes people rested.
Approximately 80% of cases of narcolepsy accompanied by cataplexy: sudden episodic loss of muscle tone that is repeated regularly. In mild cases, the patient's lower jaw droops slightly, and there is a feeling of weakness in the knees, but if a serious condition, the person may suddenly fall out of the blue. His mind, however, remains unclear. Cataplexy is developed on the background of pronounced emotional reactions: laughter, anger, fear or surprise that makes this state is particularly inconvenient.
The cause of the disease is not completely clear, but in some cases, there is a mutation of the neurotransmitter orexin (gene HCRT, 17q21), which regulates the transfer of excitation signals in the brain and affects sleep and appetite. The signaling system between oreksinergicheskimi and other neurons fails, inhibits the activity of monoaminergic neurons and reduced inflow of excitatory signals to the cortex.
Eliminate narcolepsy-cataplexy is not possible, however, symptomatic treatment is. Patients begin to feel better through regular sleep at a fixed time and drugs, activates the central adrenergic systems.
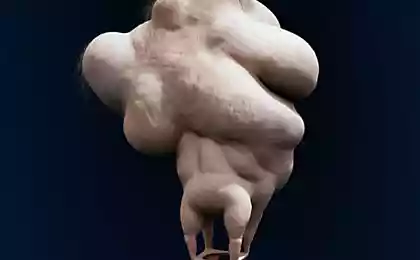
Fibrodisplaziya
Fibrodisplations ossificans progressiva (FOP) - a rare genetic disorder in which the body starts to generate new bone - ossification - in the wrong places: in muscles, ligaments, tendons and other connective tissues. These can cause the formation of any trauma: bruises, cuts, broken, intramuscular injection or surgery. Because of this it is impossible to remove ossification: after surgical bone can only grow stronger. Physiologically ossification do not differ from ordinary bone and can withstand heavy loads, here are just not where it should be.
PVD occurs due to a mutation in the gene ACVR1 / ALK2, coding for a bone morphogenetic protein receptor. It is transmitted to humans inherited from one parent, if he is sick, too. Being a carrier of the disease can not be: the patient is either sick or not. While FOP is one of incurable diseases, but now a second series of tests of the drug under the name palovaroten that allows you to lock the gene responsible for the pathology.
Progeriya
Children Progeria, or Hutchinson-Gilford syndrome - a disease that affects one person out of four to seven million. In children with this diagnosis is aging very rapidly and early: as a teenager patients look and feel like an old man. They develop a lot of senile pathologies lead to abnormalities in the internal organs and systems, bones, skin, muscles and tendons become weak and lethargic. At the same time the level of development of children with progeria are not inferior to their peers, and sometimes ahead of them. The average life expectancy of people suffering from the syndrome of Hutchinson-Gilford - 13 years. As a rule, it becomes a cause of death of myocardial infarction. Described only one case, when a patient with this diagnosis live up to 45 years.
The cause of the children's progeria - a spontaneous mutation in one of two copies (alleles) of the gene LMNA, encoding prelamin, A, which is a precursor to the mature forms of lamins A and C. These proteins are required for the shell of the cell nuclei were intact and functioning normally. Mutant prelamin, A - or "progerin", as suggested by some authors call it - is accumulated in the cells, so that their nuclear shell shrink and become irregular shape of the nucleus. These cells can no longer divide normally, so that the body loses its ability to not only grow, but also to replace the dying cells with new.
Currently, the disease is also considered incurable, but extend the life of patients and helps improve the quality of a variety of symptomatic treatment and physical activity, especially swimming. These tools can improve the circulatory system and joints. In addition, growth hormone is used.
Syndrome ROHHAD
ROHHAD Syndrome (Rapid-onset Obesity with Hypothalamic dysfunction, Hypoventilation and Autonomic Dysregulation) - an extremely rare disease in which a person begins to rapidly gain weight and suffer from bulimia, respiratory diseases, pauses in breathing during sleep, alveolar hypoventilation, and cardiorespiratory stops. Also in patients with diagnosed characteristic lack of response to increasing levels of carbon dioxide.
Today in the world registered about 100 cases of this disorder. Usually it appears in the age of 10 (usually about 3 years), and apparently has a hereditary nature. Although studies conducted in the West, the etiology of the syndrome ROHHAD still not clear. It is believed that it arises from the dysfunction of the pituitary, which cause genetic mutations. However, scientists have yet to determine which process is violated in this case.

fatal familial insomnia, children's age and the extra bone in the wrong places - it's not only the ideas, inspirational filmmakers, but also real hereditary diseases that occur as a result of genetic mutations. "Theory and Practice" compiled a list of the most unimaginable diseases and describe their causes.
Fatal familial bessonnitsa

Fatal familial insomnia - a rare hereditary disease in which a person dies from an inability to sleep. Until now, it was observed only forty families around the world. Fatal Insomnia usually manifests between 30 and 60 years (usually - after 50 years) and extends from 7 to 36 months. As the disease progresses, the patient suffers from a more serious sleep disorders, and no sleeping pills do not help him. The first stage is accompanied by insomnia, panic attacks and phobias, the second added to them hallucinations and increased sweating. At the third stage of the disease the person loses the ability to sleep and begins to look much older than his years. Then developing dementia, and the patient dies - usually from starvation or pneumonia.
Lethal insomnia arises from the fact that in a codon (encoding trinucleotide) 178 gene PRNP, located at 20 chromosome instead of asparagine, which is normally present in the body structure of proteins and is required for normal functioning of the nervous system, appears aspartic acid. This changes the shape of the protein molecule, and it turns into a prion - aggressive abnormal protein in the composition which have nucleic acids. Under the influence of the surrounding prion molecules become like him, and this leads to irreversible changes. Before that, they occur in the tissues of the thalamus: subcortical "stations" of all kinds of sensitivity, including responsible for the occurrence of somnolence and motor functions: swallowing, sucking, chewing, laughing. Under the action of prions thalamic nuclei covered pores turn into a sponge and stop working.
The disease is characterized by an autosomal dominant mode of inheritance: that is, it does not have media. To children it is transmitted from parents with a probability of 50%, and only on condition that one of them is sick. Men and women suffer from fatal familial insomnia with the same frequency. Today the disease is considered incurable.
Narcolepsy-katapleksiya

Narcolepsy-cataplexy syndrome, which is characterized by sudden onset of sleep and relaxation of the muscles of the body, too, has a genetic nature and arises from the rapid phase of sleep disorders. It occurs more often fatal familial insomnia: at forty out of every one hundred thousand people, equally men and women. A person suffering from narcolepsy, suddenly able to sleep for a few minutes in the middle of the day. "Sleepy attacks" remind REM sleep and can happen very often - up to a hundred times a day, preceded by a headache or without. They are often triggered by inactivity, but can occur at exactly the wrong time during intercourse, sports, driving. Wakes people rested.
Approximately 80% of cases of narcolepsy accompanied by cataplexy: sudden episodic loss of muscle tone that is repeated regularly. In mild cases, the patient's lower jaw droops slightly, and there is a feeling of weakness in the knees, but if a serious condition, the person may suddenly fall out of the blue. His mind, however, remains unclear. Cataplexy is developed on the background of pronounced emotional reactions: laughter, anger, fear or surprise that makes this state is particularly inconvenient.
The cause of the disease is not completely clear, but in some cases, there is a mutation of the neurotransmitter orexin (gene HCRT, 17q21), which regulates the transfer of excitation signals in the brain and affects sleep and appetite. The signaling system between oreksinergicheskimi and other neurons fails, inhibits the activity of monoaminergic neurons and reduced inflow of excitatory signals to the cortex.
Eliminate narcolepsy-cataplexy is not possible, however, symptomatic treatment is. Patients begin to feel better through regular sleep at a fixed time and drugs, activates the central adrenergic systems.
Fibrodisplaziya

Fibrodisplations ossificans progressiva (FOP) - a rare genetic disorder in which the body starts to generate new bone - ossification - in the wrong places: in muscles, ligaments, tendons and other connective tissues. These can cause the formation of any trauma: bruises, cuts, broken, intramuscular injection or surgery. Because of this it is impossible to remove ossification: after surgical bone can only grow stronger. Physiologically ossification do not differ from ordinary bone and can withstand heavy loads, here are just not where it should be.
PVD occurs due to a mutation in the gene ACVR1 / ALK2, coding for a bone morphogenetic protein receptor. It is transmitted to humans inherited from one parent, if he is sick, too. Being a carrier of the disease can not be: the patient is either sick or not. While FOP is one of incurable diseases, but now a second series of tests of the drug under the name palovaroten that allows you to lock the gene responsible for the pathology.
Progeriya

Children Progeria, or Hutchinson-Gilford syndrome - a disease that affects one person out of four to seven million. In children with this diagnosis is aging very rapidly and early: as a teenager patients look and feel like an old man. They develop a lot of senile pathologies lead to abnormalities in the internal organs and systems, bones, skin, muscles and tendons become weak and lethargic. At the same time the level of development of children with progeria are not inferior to their peers, and sometimes ahead of them. The average life expectancy of people suffering from the syndrome of Hutchinson-Gilford - 13 years. As a rule, it becomes a cause of death of myocardial infarction. Described only one case, when a patient with this diagnosis live up to 45 years.
The cause of the children's progeria - a spontaneous mutation in one of two copies (alleles) of the gene LMNA, encoding prelamin, A, which is a precursor to the mature forms of lamins A and C. These proteins are required for the shell of the cell nuclei were intact and functioning normally. Mutant prelamin, A - or "progerin", as suggested by some authors call it - is accumulated in the cells, so that their nuclear shell shrink and become irregular shape of the nucleus. These cells can no longer divide normally, so that the body loses its ability to not only grow, but also to replace the dying cells with new.
Currently, the disease is also considered incurable, but extend the life of patients and helps improve the quality of a variety of symptomatic treatment and physical activity, especially swimming. These tools can improve the circulatory system and joints. In addition, growth hormone is used.
Syndrome ROHHAD

ROHHAD Syndrome (Rapid-onset Obesity with Hypothalamic dysfunction, Hypoventilation and Autonomic Dysregulation) - an extremely rare disease in which a person begins to rapidly gain weight and suffer from bulimia, respiratory diseases, pauses in breathing during sleep, alveolar hypoventilation, and cardiorespiratory stops. Also in patients with diagnosed characteristic lack of response to increasing levels of carbon dioxide.
Today in the world registered about 100 cases of this disorder. Usually it appears in the age of 10 (usually about 3 years), and apparently has a hereditary nature. Although studies conducted in the West, the etiology of the syndrome ROHHAD still not clear. It is believed that it arises from the dysfunction of the pituitary, which cause genetic mutations. However, scientists have yet to determine which process is violated in this case.
Author: Natalia Kienya
Source: T & P i>
blockquote>
via factroom.ru