886
5 patologías genéticas humanas más inimaginables
A veces, el mismo es ... 0,
insomnio familiar fatal, la edad de los niños y el hueso extra en los lugares equivocados - es no sólo las ideas, cineastas de inspiración, sino también enfermedades hereditarias reales que se producen como resultado de mutaciones genéticas. "Teoría y Práctica" compilaron una lista de las enfermedades más inimaginables y describir sus causas.
Fatal
bessonnitsa familiar
familiar
Fatal insomnio familiar - una enfermedad hereditaria rara en la que una persona muere a causa de una incapacidad para dormir. Hasta ahora, se observó sólo cuarenta familias de todo el mundo. Fatal El insomnio suele manifestarse entre los 30 y los 60 años (por lo general - después de 50 años) y se extiende desde 7 a 36 meses. A medida que la enfermedad progresa, el paciente sufre de una más graves trastornos del sueño, y no hay pastillas para dormir no lo ayudara. La primera etapa se acompaña de insomnio, ataques de pánico y fobias, el segundo añadido a ellos alucinaciones y aumento de la sudoración. En la tercera etapa de la enfermedad la persona pierde la capacidad de dormir y comienza a mirar mucho más viejo que sus años. Entonces desarrollar demencia, y el paciente muere. - Por lo general de la inanición o neumonía
Insomnio Lethal surge del hecho de que en un codón (trinucleótido codificación) 178 PRNP gen, localizado en 20 cromosomas en lugar de asparagina, que normalmente está presente en la estructura del cuerpo de las proteínas y se requiere para el funcionamiento normal del sistema nervioso, parece ácido aspártico. Esto cambia la forma de la molécula de proteína, y se convierte en un prión - proteína anormal agresiva en la composición que tienen los ácidos nucleicos. Bajo la influencia de las moléculas priónicas que rodean a ser como él, y esto conduce a cambios irreversibles. Antes de eso, se producen en los tejidos del tálamo: "estaciones" subcorticales de todo tipo de sensibilidad, entre ellos el responsable de la aparición de somnolencia y funciones motoras: la deglución, chupados, mascados, riendo. Bajo la acción de los priones núcleos talámicos cubiertos poros se convierten en una esponja y dejan de funcionar.
La enfermedad se caracteriza por un modo de herencia autosómico dominante: es decir, que no tiene los medios de comunicación. Para los niños que se transmite de padres con una probabilidad del 50%, y sólo con la condición de que uno de ellos está enfermo. Los hombres y las mujeres sufren de insomnio familiar fatal con la misma frecuencia. Hoy en día la enfermedad se considera incurable.
Narcolepsia-katapleksiya
Síndrome de narcolepsia-cataplejía, que se caracteriza por la aparición súbita de sueño y la relajación de los músculos del cuerpo, también, tiene una naturaleza genética y surge de la fase rápida de trastornos del sueño. Es más frecuente el insomnio familiar fatal: a los cuarenta de cada cien mil personas, por igual hombres y mujeres. Una persona que sufre de narcolepsia, de repente capaz de dormir durante unos minutos en la mitad del día. "Los ataques Sleepy" recuerdan el sueño REM y pueden ocurrir muy a menudo - hasta cien veces al día, precedido por un dolor de cabeza o sin ella. A menudo son provocados por la inactividad, pero pueden ocurrir exactamente en el momento equivocado durante el coito, el deporte, la conducción. Despierta pueblo reposó.
Aproximadamente el 80% de los casos de narcolepsia acompañados de cataplejia: repentina pérdida episódica del tono muscular que se repite regularmente. En los casos leves, la mandíbula inferior del paciente se inclina ligeramente, y hay una sensación de debilidad en las rodillas, pero si una enfermedad grave, la persona puede caer repentinamente de la nada. Su mente, sin embargo, aún no está claro. La cataplejía se desarrolla en el fondo de las reacciones emocionales pronunciadas:. La risa, ira, miedo o sorpresa que hace que este estado es particularmente inconveniente
La causa de la enfermedad no es completamente clara, pero en algunos casos, hay una mutación del neurotransmisor orexina (HCRT gen, 17q21), que regula la transferencia de señales de excitación en el cerebro y afecta el sueño y el apetito. El sistema de señalización entre oreksinergicheskimi y otras neuronas falla, inhibe la actividad de las neuronas monoaminérgicos y menor flujo de señales excitadoras a la corteza.
Elimine la narcolepsia-cataplejía no es posible, sin embargo, el tratamiento sintomático es. Los pacientes comienzan a sentirse mejor a través de sueño regular a una hora fija y las drogas, activa los sistemas adrenérgicos centrales.
Fibrodisplaziya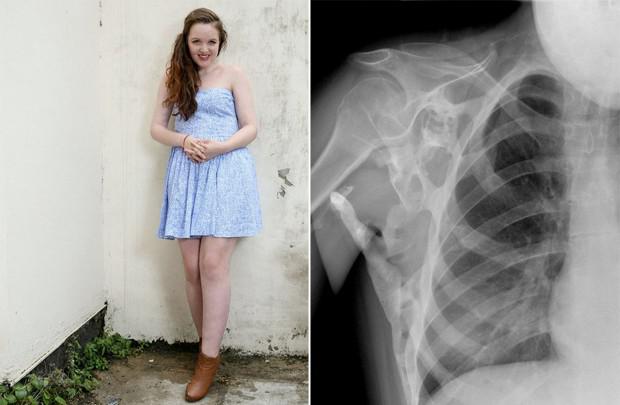
Fibrodisplations osificante progresiva (FOP) - un trastorno genético poco común en la que el cuerpo comienza a generar hueso nuevo - osificación - en los lugares equivocados: en los músculos, ligamentos, tendones y otros tejidos conectivos. Estos pueden causar la formación de cualquier trauma: contusiones, cortes,, inyección o cirugía intramuscular roto. Debido a esto, es imposible eliminar la osificación: después de hueso quirúrgico sólo puede crecer más fuerte. Osificación Fisiológicamente no difieren de hueso normal y puede soportar cargas pesadas, aquí no es donde debe estar.
PVD se produce debido a una mutación en el gen ACVR1 / ALK2, que codifica para un receptor de la proteína morfogenética ósea. Se transmite a los humanos heredados de uno de los padres, si está enfermo, también. Ser portador de la enfermedad no puede ser: el paciente está enfermo o no. Mientras FOP es una de las enfermedades incurables, pero ahora una segunda serie de pruebas de la droga bajo el nombre palovaroten que le permite bloquear el gen responsable de la patología.
Progeriya
Niños progeria o síndrome de Hutchinson-Gilford - una enfermedad que afecta a una persona fuera de cuatro para siete millones. En los niños con este diagnóstico está envejeciendo muy rápidamente y principios: como pacientes adolescentes ven y se sienten como un anciano. Desarrollan una gran cantidad de patologías seniles conducen a anormalidades en los órganos internos y sistemas, los huesos, la piel, los músculos y los tendones se debilitan y letárgico. Al mismo tiempo, el nivel de desarrollo de los niños con progeria no son inferiores a sus compañeros, y, a veces por delante de ellos. La esperanza media de vida de las personas que sufren de síndrome de Hutchinson-Gilford - 13 años. Como regla general, se convierte en una causa de la muerte de infarto de miocardio. Descrito sólo un caso, cuando un paciente con este diagnóstico viven hasta 45 años.
La causa de la progeria de los niños - una mutación espontánea en una de las dos copias (alelos) del gen LMNA, prelamin codificación, A, que es un precursor de las formas maduras de las laminas A y C. Estas proteínas son necesarias para la cáscara de los núcleos de las células estaban intactas y funcionando normalmente. Prelamin Mutante, A - o "progerina", según lo sugerido por algunos autores llaman - se acumula en las células, por lo que su cáscara nuclear encoge y se convierten en forma irregular del núcleo. Estas células ya no pueden dividir normalmente, de modo que el cuerpo pierde su capacidad de crecer no sólo, sino también para reemplazar las células que mueren con nueva.
Actualmente, la enfermedad también se considera incurable, pero a extender la vida de los pacientes y ayuda a mejorar la calidad de una variedad de tratamiento sintomático y la actividad física, especialmente la natación. Estas herramientas pueden mejorar el sistema circulatorio y las articulaciones. Además, se utiliza la hormona del crecimiento.
Síndrome ROHHAD
Síndrome ROHHAD (obesidad rápido inicio con la disfunción hipotalámica, hipoventilación y autonómica desregulación) - una enfermedad extremadamente rara en la que una persona comienza a ganar peso rápidamente y sufren de bulimia, enfermedades respiratorias, pausas en la respiración durante el sueño, hipoventilación alveolar y paradas cardiorrespiratorias. También en pacientes con diagnóstico de característica falta de respuesta a los crecientes niveles de dióxido de carbono.
Hoy en día en el mundo registró cerca de 100 casos de este trastorno. Por lo general, aparece en la edad de 10 (por lo general alrededor de 3 años), y al parecer tiene un carácter hereditario. Aunque los estudios conducidos en Occidente, la etiología del síndrome ROHHAD todavía no está claro. Se cree que surge de la disfunción de la hipófisis, lo que causa mutaciones genéticas. Sin embargo, los científicos aún tienen que determinar qué proceso se violó en este caso.

insomnio familiar fatal, la edad de los niños y el hueso extra en los lugares equivocados - es no sólo las ideas, cineastas de inspiración, sino también enfermedades hereditarias reales que se producen como resultado de mutaciones genéticas. "Teoría y Práctica" compilaron una lista de las enfermedades más inimaginables y describir sus causas.
Fatal
bessonnitsa
 familiar
familiar
Fatal insomnio familiar - una enfermedad hereditaria rara en la que una persona muere a causa de una incapacidad para dormir. Hasta ahora, se observó sólo cuarenta familias de todo el mundo. Fatal El insomnio suele manifestarse entre los 30 y los 60 años (por lo general - después de 50 años) y se extiende desde 7 a 36 meses. A medida que la enfermedad progresa, el paciente sufre de una más graves trastornos del sueño, y no hay pastillas para dormir no lo ayudara. La primera etapa se acompaña de insomnio, ataques de pánico y fobias, el segundo añadido a ellos alucinaciones y aumento de la sudoración. En la tercera etapa de la enfermedad la persona pierde la capacidad de dormir y comienza a mirar mucho más viejo que sus años. Entonces desarrollar demencia, y el paciente muere. - Por lo general de la inanición o neumonía
Insomnio Lethal surge del hecho de que en un codón (trinucleótido codificación) 178 PRNP gen, localizado en 20 cromosomas en lugar de asparagina, que normalmente está presente en la estructura del cuerpo de las proteínas y se requiere para el funcionamiento normal del sistema nervioso, parece ácido aspártico. Esto cambia la forma de la molécula de proteína, y se convierte en un prión - proteína anormal agresiva en la composición que tienen los ácidos nucleicos. Bajo la influencia de las moléculas priónicas que rodean a ser como él, y esto conduce a cambios irreversibles. Antes de eso, se producen en los tejidos del tálamo: "estaciones" subcorticales de todo tipo de sensibilidad, entre ellos el responsable de la aparición de somnolencia y funciones motoras: la deglución, chupados, mascados, riendo. Bajo la acción de los priones núcleos talámicos cubiertos poros se convierten en una esponja y dejan de funcionar.
La enfermedad se caracteriza por un modo de herencia autosómico dominante: es decir, que no tiene los medios de comunicación. Para los niños que se transmite de padres con una probabilidad del 50%, y sólo con la condición de que uno de ellos está enfermo. Los hombres y las mujeres sufren de insomnio familiar fatal con la misma frecuencia. Hoy en día la enfermedad se considera incurable.
Narcolepsia-katapleksiya

Síndrome de narcolepsia-cataplejía, que se caracteriza por la aparición súbita de sueño y la relajación de los músculos del cuerpo, también, tiene una naturaleza genética y surge de la fase rápida de trastornos del sueño. Es más frecuente el insomnio familiar fatal: a los cuarenta de cada cien mil personas, por igual hombres y mujeres. Una persona que sufre de narcolepsia, de repente capaz de dormir durante unos minutos en la mitad del día. "Los ataques Sleepy" recuerdan el sueño REM y pueden ocurrir muy a menudo - hasta cien veces al día, precedido por un dolor de cabeza o sin ella. A menudo son provocados por la inactividad, pero pueden ocurrir exactamente en el momento equivocado durante el coito, el deporte, la conducción. Despierta pueblo reposó.
Aproximadamente el 80% de los casos de narcolepsia acompañados de cataplejia: repentina pérdida episódica del tono muscular que se repite regularmente. En los casos leves, la mandíbula inferior del paciente se inclina ligeramente, y hay una sensación de debilidad en las rodillas, pero si una enfermedad grave, la persona puede caer repentinamente de la nada. Su mente, sin embargo, aún no está claro. La cataplejía se desarrolla en el fondo de las reacciones emocionales pronunciadas:. La risa, ira, miedo o sorpresa que hace que este estado es particularmente inconveniente
La causa de la enfermedad no es completamente clara, pero en algunos casos, hay una mutación del neurotransmisor orexina (HCRT gen, 17q21), que regula la transferencia de señales de excitación en el cerebro y afecta el sueño y el apetito. El sistema de señalización entre oreksinergicheskimi y otras neuronas falla, inhibe la actividad de las neuronas monoaminérgicos y menor flujo de señales excitadoras a la corteza.
Elimine la narcolepsia-cataplejía no es posible, sin embargo, el tratamiento sintomático es. Los pacientes comienzan a sentirse mejor a través de sueño regular a una hora fija y las drogas, activa los sistemas adrenérgicos centrales.
Fibrodisplaziya

Fibrodisplations osificante progresiva (FOP) - un trastorno genético poco común en la que el cuerpo comienza a generar hueso nuevo - osificación - en los lugares equivocados: en los músculos, ligamentos, tendones y otros tejidos conectivos. Estos pueden causar la formación de cualquier trauma: contusiones, cortes,, inyección o cirugía intramuscular roto. Debido a esto, es imposible eliminar la osificación: después de hueso quirúrgico sólo puede crecer más fuerte. Osificación Fisiológicamente no difieren de hueso normal y puede soportar cargas pesadas, aquí no es donde debe estar.
PVD se produce debido a una mutación en el gen ACVR1 / ALK2, que codifica para un receptor de la proteína morfogenética ósea. Se transmite a los humanos heredados de uno de los padres, si está enfermo, también. Ser portador de la enfermedad no puede ser: el paciente está enfermo o no. Mientras FOP es una de las enfermedades incurables, pero ahora una segunda serie de pruebas de la droga bajo el nombre palovaroten que le permite bloquear el gen responsable de la patología.
Progeriya

Niños progeria o síndrome de Hutchinson-Gilford - una enfermedad que afecta a una persona fuera de cuatro para siete millones. En los niños con este diagnóstico está envejeciendo muy rápidamente y principios: como pacientes adolescentes ven y se sienten como un anciano. Desarrollan una gran cantidad de patologías seniles conducen a anormalidades en los órganos internos y sistemas, los huesos, la piel, los músculos y los tendones se debilitan y letárgico. Al mismo tiempo, el nivel de desarrollo de los niños con progeria no son inferiores a sus compañeros, y, a veces por delante de ellos. La esperanza media de vida de las personas que sufren de síndrome de Hutchinson-Gilford - 13 años. Como regla general, se convierte en una causa de la muerte de infarto de miocardio. Descrito sólo un caso, cuando un paciente con este diagnóstico viven hasta 45 años.
La causa de la progeria de los niños - una mutación espontánea en una de las dos copias (alelos) del gen LMNA, prelamin codificación, A, que es un precursor de las formas maduras de las laminas A y C. Estas proteínas son necesarias para la cáscara de los núcleos de las células estaban intactas y funcionando normalmente. Prelamin Mutante, A - o "progerina", según lo sugerido por algunos autores llaman - se acumula en las células, por lo que su cáscara nuclear encoge y se convierten en forma irregular del núcleo. Estas células ya no pueden dividir normalmente, de modo que el cuerpo pierde su capacidad de crecer no sólo, sino también para reemplazar las células que mueren con nueva.
Actualmente, la enfermedad también se considera incurable, pero a extender la vida de los pacientes y ayuda a mejorar la calidad de una variedad de tratamiento sintomático y la actividad física, especialmente la natación. Estas herramientas pueden mejorar el sistema circulatorio y las articulaciones. Además, se utiliza la hormona del crecimiento.
Síndrome ROHHAD

Síndrome ROHHAD (obesidad rápido inicio con la disfunción hipotalámica, hipoventilación y autonómica desregulación) - una enfermedad extremadamente rara en la que una persona comienza a ganar peso rápidamente y sufren de bulimia, enfermedades respiratorias, pausas en la respiración durante el sueño, hipoventilación alveolar y paradas cardiorrespiratorias. También en pacientes con diagnóstico de característica falta de respuesta a los crecientes niveles de dióxido de carbono.
Hoy en día en el mundo registró cerca de 100 casos de este trastorno. Por lo general, aparece en la edad de 10 (por lo general alrededor de 3 años), y al parecer tiene un carácter hereditario. Aunque los estudios conducidos en Occidente, la etiología del síndrome ROHHAD todavía no está claro. Se cree que surge de la disfunción de la hipófisis, lo que causa mutaciones genéticas. Sin embargo, los científicos aún tienen que determinar qué proceso se violó en este caso.
Autor: Natalia Kienya
Fuente: T & P i>
Blockquote>
a través factroom.ru