886
5 найбільш генетичних патологій людини
Що відбувається. . 
Жирні сімейні безсоння, дитячий вік і надлишок кісток в неправильному місці – це не тільки ідеї, які надихають кінематографістів, але і реальні спадкові захворювання, які виникають в результаті генетичних мутацій. «Теорія і практика» скомпільували список найбільш незліковних захворювань і описали причини їх виникнення.
Жирна фоміальна безсоння
Жирна фміліальна безсоння є рідкісним спадковим захворюванням, в якому людина гине від нездатності падати. На сьогоднішній день вона відсвяткувала тільки сім'ї, які проживають у всьому світі. Жирна безсоння зазвичай виникає між 30 і 60 років (понад 50 років) і триває від 7 до 36 місяців. Як хвороба прогресує, хворий страждає від більш серйозних порушень сну, і не спить таблетки допомогти йому. У першому етапі інсомнія супроводжується панічними атаками і фобійцями, в другому, до них додаються галюцинації і підвищені потовиділення. На третьому етапі захворювання людина повністю втрачає здатність спати і починає виглядати набагато старше років. Після чого дементія розвивається і хворі гинуть, зазвичай від виснаження або пневмонії.
Жирна безсоння виникає через те, що в кодоні (кодування тринуклеотиду) 178 гена PRNP розташований на 20-му хромосомі, замість аспагіну, який зазвичай присутній в організмі в складі білків і необхідний для нормального функціонування нервової системи, з'являється апартічна кислота. Це змінює форму молекули білка, і вона перетворюється в пропі, агресивний аномальний білок, який містить не нуклеїнові кислоти. Під дією однієї обрізки, навколишні молекули подобаються їй, і це призводить до незворотних змін. Перед тим, що виникають в тканинах таламуса: підкортна «станція» всіх видів чутливості, відповідальна за виникнення сонного стану і моторних функцій: поковтання, смоктання, жування, сміхання. Під дією ріпчастих нуклейок таламуса покривають пори, перетворюються на губку і зупиняються роботи.
Захворювання характеризується автоматично домінантним типом спадщини: тобто не має носіїв. Передається дітям з батьків з ймовірністю 50% і тільки якщо один з них хворий. Чоловіки і жінки страждають від жирової нездужання з однаковою частотою. На сьогоднішній день це захворювання вважається невиліковним.
Narcolepsy-cataplexy
Нарколепсій-катаплексний синдром, який характеризується раптовими нападами сну і розслабленням м'язів тіла, також має генетичну природу і виникає через порушення швидкої фази сну. Настає багато частіше жирної сім'ї безсоння: в сорокі від сто тисяч людей, однаково у чоловіків і жінок. Людина, яка страждає від narcolepsy, може раптово закохати протягом декількох хвилин в середині дня. "Подарує атаки" нагадують фазу сну РЕМ і може статися дуже часто - до сотні разів на день, з або без головного болю перед ними. Вони часто спровоковані бездіяльністю, але можуть виникати в абсолютно невідповідний час: під час статевого акту, спорту, водіння. Людина прокидається відпочивати.
Приблизно 80% випадків narcolepsy супроводжуються катаплексією: раптовою епісоціальною втратою тонусу м'язів, яка регулярно проводить. У м'яких випадках хворий злегка відвисає нижню щелепу і виникає відчуття слабкості в колінах, але якщо стан сильний, людина може раптово попадати на плоске місце. Його свідомість залишається чіткою. Катаплексія розвивається на тлі виражених емоційних реакцій: сміху, гніву, страху або сюрпризу, що робить цей стан особливо несприятливим.
Причина захворювання ще не зрозуміло, але в деяких випадках виникла мутація нейротрансміттера абоексину (ГКРТ ген, 17q21), яка регулює передачу збудливих сигналів в мозку і впливає на сон і апетит. Пригнічують систему сигналізації між анексинергією та іншими нейронами, активність моноамінергічних нейронів та інфлюкс збудливих сигналів у кору.
Неможливе усунення narcolepsy-cataplexy, але можливе лікування симптоматики. Пацієнти починають відчувати себе краще за рахунок регулярного сну в певний час і лікарських засобів, які активують центральні адренергічні системи.
Фібродисплазія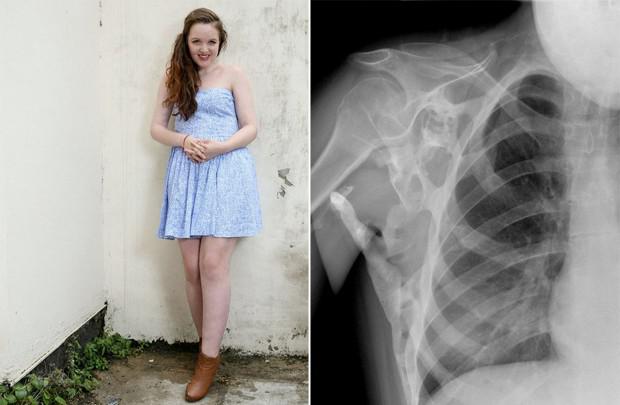
Озування прогресивної фіброзлазії (FOP) є рідкісною генетичною хворобою, в якій організм починає формувати нові кістки - оситивні засоби - в неправильних місцях: всередині м'язів, зв'язок, сухожилля та інших сполучних тканин. Будь-яка травма може призвести до їх утворення: синус, ріжучий, перелом, внутрішньом'язова ін'єкції або операції. Через це неможливо прибрати зозів: після операції кістки може тільки сильніше рости. Фізіологічно не відрізняються від звичайних кісток і можуть витримати значні навантаження, тільки вони не повинні бути.
FOP виникає через мутація в гені ACVR1 / ALK2, що кодує рецептор морфогенетичного білка. Пройшов на людину, спадаючи від одного з батьків, якщо він також хворий. Не можна бути носієм цього захворювання: хворий або не хворий. В той час як FOP є одним з невиліковних захворювань, але зараз другий ряд тестів препарату називається паловотен, що дозволяє блокувати ген, відповідальний за патологію.
Прогерія
Прогерія для дітей або синдром Hutchinson-Gilford - це захворювання, що впливає на однечотирьох до семи мільйонів людей. Тіло дітей з цим діагнозом дуже швидко і рано: вже в підлітковому віці хворі виглядають і відчувають себе як старі люди. Розвивають багато генетичних патологій, виникають порушення в роботі внутрішніх органів і систем, і кісток, шкіри, м'язів і сухожилля стають слабкими і ламкими. У той же час діти з прогерією не поступаються своїм одноліткам в плані розвитку, а іноді перед ними. Середня тривалість життя людей з синдромом Hutchinson-Gilford становить 13 років. Як правило, причиною смерті є інфаркт міокарда. Тільки один випадок був описаний, коли хворий з такою діагнозом жив до 45 років.
Причиною розвитку дитячої прогерії є спонтанна мутація в одному з двох примірників (алелей) гена LMNA, преламін, А, який є прекурсором зрілих форм lamines, A і C. Ці білки необхідні для оболонки нуклеуса клітинки, щоб бути цілими і функціонувати нормально. мутантний преламин, А— або «прогерін», як і деякі автори пропонують, накопичуються в клітинах, щоб їх ядерні оболонки рятують і їх нуклеї приймають на нерегулярну форму. Такі клітини більше не можна розділити нормально, тому організм втрачає здатність не тільки виростити, але і замінити дисперсні клітини новими.
На даний момент це захворювання вважається невиліковним, але для продовження життя пацієнтів і поліпшення його якості допомагає різноманітність симптоматичного лікування і фізичної активності, особливо плавання. Ці кошти можуть поліпшити стан системи кровообігу та суглобів. Також використовується гормон росту.
Синдром Рогад
Короткострокова ожиріння з дисфункцією гіповентилами, гіповентилацією та автономічними дисрегуляціями є надзвичайно рідкісним захворюванням, при якому людина починає швидко набирати вагу і страждає від булімії, респіраторних захворювань, затримок дихання сну, альвеолярної гіповентиляції та кардіореспіраторних арештів. Також хворі з такою діагностикою характеризуються відсутністю реакції на збільшення вуглекислого газу в крові.
На сьогоднішній день існує близько 100 випадків цього розладу у світі. Як правило, з'являється до 10-ти років (найчастіше близько 3 років) і, очевидно, має спадкову природу. Незважаючи на західні дослідження, етіологія синдрому Rohhad ще не зрозуміло. Вважається, що виникає через дисфункцію гіпофіза, що викликається генетичною мутацією. Однак вчені ще вирішили визначити, який процес порушується в цьому випадку.
Автор: Наталія Кєня
Джерело: T&P
Веб-камера

Жирні сімейні безсоння, дитячий вік і надлишок кісток в неправильному місці – це не тільки ідеї, які надихають кінематографістів, але і реальні спадкові захворювання, які виникають в результаті генетичних мутацій. «Теорія і практика» скомпільували список найбільш незліковних захворювань і описали причини їх виникнення.
Жирна фоміальна безсоння

Жирна фміліальна безсоння є рідкісним спадковим захворюванням, в якому людина гине від нездатності падати. На сьогоднішній день вона відсвяткувала тільки сім'ї, які проживають у всьому світі. Жирна безсоння зазвичай виникає між 30 і 60 років (понад 50 років) і триває від 7 до 36 місяців. Як хвороба прогресує, хворий страждає від більш серйозних порушень сну, і не спить таблетки допомогти йому. У першому етапі інсомнія супроводжується панічними атаками і фобійцями, в другому, до них додаються галюцинації і підвищені потовиділення. На третьому етапі захворювання людина повністю втрачає здатність спати і починає виглядати набагато старше років. Після чого дементія розвивається і хворі гинуть, зазвичай від виснаження або пневмонії.
Жирна безсоння виникає через те, що в кодоні (кодування тринуклеотиду) 178 гена PRNP розташований на 20-му хромосомі, замість аспагіну, який зазвичай присутній в організмі в складі білків і необхідний для нормального функціонування нервової системи, з'являється апартічна кислота. Це змінює форму молекули білка, і вона перетворюється в пропі, агресивний аномальний білок, який містить не нуклеїнові кислоти. Під дією однієї обрізки, навколишні молекули подобаються їй, і це призводить до незворотних змін. Перед тим, що виникають в тканинах таламуса: підкортна «станція» всіх видів чутливості, відповідальна за виникнення сонного стану і моторних функцій: поковтання, смоктання, жування, сміхання. Під дією ріпчастих нуклейок таламуса покривають пори, перетворюються на губку і зупиняються роботи.
Захворювання характеризується автоматично домінантним типом спадщини: тобто не має носіїв. Передається дітям з батьків з ймовірністю 50% і тільки якщо один з них хворий. Чоловіки і жінки страждають від жирової нездужання з однаковою частотою. На сьогоднішній день це захворювання вважається невиліковним.
Narcolepsy-cataplexy

Нарколепсій-катаплексний синдром, який характеризується раптовими нападами сну і розслабленням м'язів тіла, також має генетичну природу і виникає через порушення швидкої фази сну. Настає багато частіше жирної сім'ї безсоння: в сорокі від сто тисяч людей, однаково у чоловіків і жінок. Людина, яка страждає від narcolepsy, може раптово закохати протягом декількох хвилин в середині дня. "Подарує атаки" нагадують фазу сну РЕМ і може статися дуже часто - до сотні разів на день, з або без головного болю перед ними. Вони часто спровоковані бездіяльністю, але можуть виникати в абсолютно невідповідний час: під час статевого акту, спорту, водіння. Людина прокидається відпочивати.
Приблизно 80% випадків narcolepsy супроводжуються катаплексією: раптовою епісоціальною втратою тонусу м'язів, яка регулярно проводить. У м'яких випадках хворий злегка відвисає нижню щелепу і виникає відчуття слабкості в колінах, але якщо стан сильний, людина може раптово попадати на плоске місце. Його свідомість залишається чіткою. Катаплексія розвивається на тлі виражених емоційних реакцій: сміху, гніву, страху або сюрпризу, що робить цей стан особливо несприятливим.
Причина захворювання ще не зрозуміло, але в деяких випадках виникла мутація нейротрансміттера абоексину (ГКРТ ген, 17q21), яка регулює передачу збудливих сигналів в мозку і впливає на сон і апетит. Пригнічують систему сигналізації між анексинергією та іншими нейронами, активність моноамінергічних нейронів та інфлюкс збудливих сигналів у кору.
Неможливе усунення narcolepsy-cataplexy, але можливе лікування симптоматики. Пацієнти починають відчувати себе краще за рахунок регулярного сну в певний час і лікарських засобів, які активують центральні адренергічні системи.
Фібродисплазія

Озування прогресивної фіброзлазії (FOP) є рідкісною генетичною хворобою, в якій організм починає формувати нові кістки - оситивні засоби - в неправильних місцях: всередині м'язів, зв'язок, сухожилля та інших сполучних тканин. Будь-яка травма може призвести до їх утворення: синус, ріжучий, перелом, внутрішньом'язова ін'єкції або операції. Через це неможливо прибрати зозів: після операції кістки може тільки сильніше рости. Фізіологічно не відрізняються від звичайних кісток і можуть витримати значні навантаження, тільки вони не повинні бути.
FOP виникає через мутація в гені ACVR1 / ALK2, що кодує рецептор морфогенетичного білка. Пройшов на людину, спадаючи від одного з батьків, якщо він також хворий. Не можна бути носієм цього захворювання: хворий або не хворий. В той час як FOP є одним з невиліковних захворювань, але зараз другий ряд тестів препарату називається паловотен, що дозволяє блокувати ген, відповідальний за патологію.
Прогерія

Прогерія для дітей або синдром Hutchinson-Gilford - це захворювання, що впливає на однечотирьох до семи мільйонів людей. Тіло дітей з цим діагнозом дуже швидко і рано: вже в підлітковому віці хворі виглядають і відчувають себе як старі люди. Розвивають багато генетичних патологій, виникають порушення в роботі внутрішніх органів і систем, і кісток, шкіри, м'язів і сухожилля стають слабкими і ламкими. У той же час діти з прогерією не поступаються своїм одноліткам в плані розвитку, а іноді перед ними. Середня тривалість життя людей з синдромом Hutchinson-Gilford становить 13 років. Як правило, причиною смерті є інфаркт міокарда. Тільки один випадок був описаний, коли хворий з такою діагнозом жив до 45 років.
Причиною розвитку дитячої прогерії є спонтанна мутація в одному з двох примірників (алелей) гена LMNA, преламін, А, який є прекурсором зрілих форм lamines, A і C. Ці білки необхідні для оболонки нуклеуса клітинки, щоб бути цілими і функціонувати нормально. мутантний преламин, А— або «прогерін», як і деякі автори пропонують, накопичуються в клітинах, щоб їх ядерні оболонки рятують і їх нуклеї приймають на нерегулярну форму. Такі клітини більше не можна розділити нормально, тому організм втрачає здатність не тільки виростити, але і замінити дисперсні клітини новими.
На даний момент це захворювання вважається невиліковним, але для продовження життя пацієнтів і поліпшення його якості допомагає різноманітність симптоматичного лікування і фізичної активності, особливо плавання. Ці кошти можуть поліпшити стан системи кровообігу та суглобів. Також використовується гормон росту.
Синдром Рогад

Короткострокова ожиріння з дисфункцією гіповентилами, гіповентилацією та автономічними дисрегуляціями є надзвичайно рідкісним захворюванням, при якому людина починає швидко набирати вагу і страждає від булімії, респіраторних захворювань, затримок дихання сну, альвеолярної гіповентиляції та кардіореспіраторних арештів. Також хворі з такою діагностикою характеризуються відсутністю реакції на збільшення вуглекислого газу в крові.
На сьогоднішній день існує близько 100 випадків цього розладу у світі. Як правило, з'являється до 10-ти років (найчастіше близько 3 років) і, очевидно, має спадкову природу. Незважаючи на західні дослідження, етіологія синдрому Rohhad ще не зрозуміло. Вважається, що виникає через дисфункцію гіпофіза, що викликається генетичною мутацією. Однак вчені ще вирішили визначити, який процес порушується в цьому випадку.
Автор: Наталія Кєня
Джерело: T&P
Веб-камера