886
5个最难以想象的人类遗传病症
有时,相同的是... 0.
致死性家族性失眠症,孩子的年龄和在错误的地方多余的骨头 - 这不仅是因为发生基因突变的结果的思想,鼓舞人心的电影制片人,也是真正的遗传性疾病。 “理论与实践”编制的最不可思议的疾病的清单,并说明其原因。
致死性家族性bessonnitsa
致死性家族性失眠症 - 一种罕见的遗传疾病,其中一个人从不能入睡死亡。到现在为止,已经观察仅40世界各地的家庭。严重失眠通常体现年30至60(通常为 - 50年后),并从7至36个月扩大。随着病情的发展,所述患者从一个更严重的睡眠障碍症,和没有安眠药不帮助他。第一阶段是伴随着失眠,惊恐发作和恐怖症,第二加到他们幻觉和出汗增多。在疾病的第三阶段的人失去睡觉的能力,并开始看起来比他多年老得多。然后患痴呆症,而且患者死亡 - 通常由饥饿或肺炎
致死失眠源于以下事实:在一个密码子(编码三核苷酸)178基因PRNP,位于20号染色体,而不是天冬酰胺,这是通常存在于蛋白质的车身结构和所需的神经系统的正常功能,出现天冬氨酸。这改变了蛋白质分子的形状,并且它变成一个朊病毒 - 侵略性异常蛋白,其中有核酸的组合物。在周围朊病毒分子的影响变得和他一样,这会导致不可逆转的变化。在此之前,他们发生在丘脑组织:皮质下“站”各种敏感的,包括负责嗜睡和运动功能发生:吞咽,吸吮,咀嚼,笑了起来。朊病毒下丘脑核覆盖毛孔变成海绵和停止工作的作用。
该疾病的特征在于继承的一个常染色体显性方式:即,它不具有媒体。对儿童是从父母以50%的概率发送,前提条件是它们中的一个是生病。男性和女性患有致死性家族性失眠症以相同的频率。今天,这种疾病被认为是不治之症。
发作性睡病,katapleksiya
嗜睡症-猝倒综合征,其特征在于突然发作睡眠身体的肌肉和放松的,也有遗传性和起因于睡眠障碍的快速相。它发生更经常致死性家族性失眠症:每40出十万人,同样男性和女性。一个人从患有嗜睡症,突然能够睡了几分钟,在一天中。 “断头攻击”提醒REM睡眠,可以很经常发生 - 高达一百次,每天,前面有一个头痛或没有。他们经常被闲置触发,但是在性交时,运动可以发生在完全错误的时间,开车。醒来的人安息。
约80%伴有猝倒发作性睡病的病例:肌张力的突然发作损失定期重复。在温和的情况下,患者的下颌稍下垂,并有弱点在膝盖的感觉,但如果病情严重,该人可能会突然掉下来的蓝色。他的心,但是,目前还不清楚。猝倒是在明显的情绪反应的背景下发展起来:笑声,愤怒,恐惧或惊讶,使这个状态是特别不方便
疾病的原因还不完全清楚,但在某些情况下,有神经递质食欲素(基因HCRT,17q21上),其中规定的激励信号的在脑中的转让和影响睡眠和食欲的突变。 oreksinergicheskimi和其他神经元失败之间的信令系统,抑制单胺能神经元的活性和兴奋性信号到皮质减少流入。
消除嗜睡症-猝倒是不可能的,但是,对症治疗。病人开始感到过有规律的睡眠更好地在固定的时间和药品,激活中央肾上腺素能系统。
Fibrodisplaziya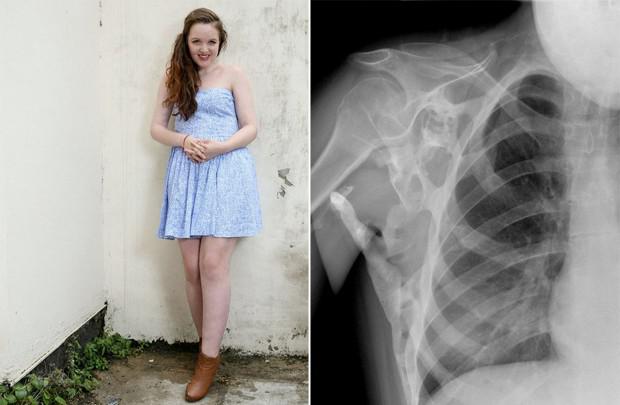
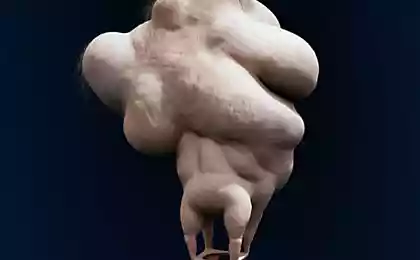
Fibrodisplations骨化渐进症(FOP) - 僵化 - - 在错误的地方在体内开始产生新骨的一种罕见的遗传性疾病:肌肉,韧带,肌腱等结缔组织。这些可能会造成任何损伤的形成:跌打损伤,刀伤,破碎,肌内注射或手术。正因为如此它是不可能消除骨化:手术后骨骼也只能变得更加强大。生理骨化不与普通骨不同,可承受重载荷,这里只是不是它应该是。
PVD的发生是由于在该基因ACVR1 / ALK2的突变,编码一种骨形态发生蛋白受体。它传染给人类从一个亲本遗传,如果他生病了。依据疾病的载体不能是:病人可以是生病或没有。虽然FOP是疑难杂症,但现在第二个系列的名义下palovaroten,使您可以锁定负责的病理基因的药物测试。
Progeriya
儿童早衰,或哈钦森 - 吉尔福特综合征 - 一种疾病,影响到一个人出四个700万。在孩子下此诊断老龄化非常迅速,早期的:作为一个十几岁的患者看起来像是一个老人。他们开发了大量的老年病症导致内部器官和系统,骨骼,皮肤,肌肉和肌腱异常变弱,昏昏欲睡。同时,早老症儿童的发展水平并不逊色于同龄人,有时在他们面前。 13年 - 人从哈钦森 - 吉尔福特的综合症的平均寿命。作为一项规则,就变成心肌梗塞死亡的原因。描述只有一种情况下,当有这种诊断病人活到45岁。
孩子们的早衰的原因 - 中的基因LMNA两个拷贝(等位基因)一种自发突变,编码prelamin,A,这是一个前体纤层蛋白A和C,这些蛋白的所需的细胞核的壳的成熟形式是完整和功能正常。突变prelamin,A - 或“早衰蛋白”建议的那样,一些学者称之为 - 积累在细胞,使它们的核壳收缩,成为核的不规则形状。这些细胞不再分裂,通常,以使身体失去它的能力不仅生长,但也以替换死亡的细胞与新。
目前,该疾病也被认为是不可治愈的,但延长患者的寿命,并有助于改善各种对症治疗和体力活动,特别是游泳的质量。这些工具可以提高循环系统和关节。另外,生长激素被使用。
综合征ROHHAD
ROHHAD综合征(发病急骤肥胖与下丘脑功能障碍,通气不足和自主神经失调) - 一,其中一人开始快速增加体重和贪食症,呼吸道疾病患极为罕见的疾病,暂停呼吸睡眠,肺泡通气不足,以及心肺功能停止时。此外,在患者的诊断特性缺乏应对二氧化碳水平的增加的。
今天,在世界上注册的约100情况下,这种障碍。它通常出现在10(一般为3年)的年龄,显然有一种遗传性的。尽管研究在西方,该综合征ROHHAD的病因尚不清楚进行。据认为,它产生于垂体功能障碍,这导致遗传突变。然而,科学家们还没有确定哪个进程被违反在这种情况下。

致死性家族性失眠症,孩子的年龄和在错误的地方多余的骨头 - 这不仅是因为发生基因突变的结果的思想,鼓舞人心的电影制片人,也是真正的遗传性疾病。 “理论与实践”编制的最不可思议的疾病的清单,并说明其原因。
致死性家族性bessonnitsa

致死性家族性失眠症 - 一种罕见的遗传疾病,其中一个人从不能入睡死亡。到现在为止,已经观察仅40世界各地的家庭。严重失眠通常体现年30至60(通常为 - 50年后),并从7至36个月扩大。随着病情的发展,所述患者从一个更严重的睡眠障碍症,和没有安眠药不帮助他。第一阶段是伴随着失眠,惊恐发作和恐怖症,第二加到他们幻觉和出汗增多。在疾病的第三阶段的人失去睡觉的能力,并开始看起来比他多年老得多。然后患痴呆症,而且患者死亡 - 通常由饥饿或肺炎
致死失眠源于以下事实:在一个密码子(编码三核苷酸)178基因PRNP,位于20号染色体,而不是天冬酰胺,这是通常存在于蛋白质的车身结构和所需的神经系统的正常功能,出现天冬氨酸。这改变了蛋白质分子的形状,并且它变成一个朊病毒 - 侵略性异常蛋白,其中有核酸的组合物。在周围朊病毒分子的影响变得和他一样,这会导致不可逆转的变化。在此之前,他们发生在丘脑组织:皮质下“站”各种敏感的,包括负责嗜睡和运动功能发生:吞咽,吸吮,咀嚼,笑了起来。朊病毒下丘脑核覆盖毛孔变成海绵和停止工作的作用。
该疾病的特征在于继承的一个常染色体显性方式:即,它不具有媒体。对儿童是从父母以50%的概率发送,前提条件是它们中的一个是生病。男性和女性患有致死性家族性失眠症以相同的频率。今天,这种疾病被认为是不治之症。
发作性睡病,katapleksiya

嗜睡症-猝倒综合征,其特征在于突然发作睡眠身体的肌肉和放松的,也有遗传性和起因于睡眠障碍的快速相。它发生更经常致死性家族性失眠症:每40出十万人,同样男性和女性。一个人从患有嗜睡症,突然能够睡了几分钟,在一天中。 “断头攻击”提醒REM睡眠,可以很经常发生 - 高达一百次,每天,前面有一个头痛或没有。他们经常被闲置触发,但是在性交时,运动可以发生在完全错误的时间,开车。醒来的人安息。
约80%伴有猝倒发作性睡病的病例:肌张力的突然发作损失定期重复。在温和的情况下,患者的下颌稍下垂,并有弱点在膝盖的感觉,但如果病情严重,该人可能会突然掉下来的蓝色。他的心,但是,目前还不清楚。猝倒是在明显的情绪反应的背景下发展起来:笑声,愤怒,恐惧或惊讶,使这个状态是特别不方便
疾病的原因还不完全清楚,但在某些情况下,有神经递质食欲素(基因HCRT,17q21上),其中规定的激励信号的在脑中的转让和影响睡眠和食欲的突变。 oreksinergicheskimi和其他神经元失败之间的信令系统,抑制单胺能神经元的活性和兴奋性信号到皮质减少流入。
消除嗜睡症-猝倒是不可能的,但是,对症治疗。病人开始感到过有规律的睡眠更好地在固定的时间和药品,激活中央肾上腺素能系统。
Fibrodisplaziya

Fibrodisplations骨化渐进症(FOP) - 僵化 - - 在错误的地方在体内开始产生新骨的一种罕见的遗传性疾病:肌肉,韧带,肌腱等结缔组织。这些可能会造成任何损伤的形成:跌打损伤,刀伤,破碎,肌内注射或手术。正因为如此它是不可能消除骨化:手术后骨骼也只能变得更加强大。生理骨化不与普通骨不同,可承受重载荷,这里只是不是它应该是。
PVD的发生是由于在该基因ACVR1 / ALK2的突变,编码一种骨形态发生蛋白受体。它传染给人类从一个亲本遗传,如果他生病了。依据疾病的载体不能是:病人可以是生病或没有。虽然FOP是疑难杂症,但现在第二个系列的名义下palovaroten,使您可以锁定负责的病理基因的药物测试。
Progeriya

儿童早衰,或哈钦森 - 吉尔福特综合征 - 一种疾病,影响到一个人出四个700万。在孩子下此诊断老龄化非常迅速,早期的:作为一个十几岁的患者看起来像是一个老人。他们开发了大量的老年病症导致内部器官和系统,骨骼,皮肤,肌肉和肌腱异常变弱,昏昏欲睡。同时,早老症儿童的发展水平并不逊色于同龄人,有时在他们面前。 13年 - 人从哈钦森 - 吉尔福特的综合症的平均寿命。作为一项规则,就变成心肌梗塞死亡的原因。描述只有一种情况下,当有这种诊断病人活到45岁。
孩子们的早衰的原因 - 中的基因LMNA两个拷贝(等位基因)一种自发突变,编码prelamin,A,这是一个前体纤层蛋白A和C,这些蛋白的所需的细胞核的壳的成熟形式是完整和功能正常。突变prelamin,A - 或“早衰蛋白”建议的那样,一些学者称之为 - 积累在细胞,使它们的核壳收缩,成为核的不规则形状。这些细胞不再分裂,通常,以使身体失去它的能力不仅生长,但也以替换死亡的细胞与新。
目前,该疾病也被认为是不可治愈的,但延长患者的寿命,并有助于改善各种对症治疗和体力活动,特别是游泳的质量。这些工具可以提高循环系统和关节。另外,生长激素被使用。
综合征ROHHAD

ROHHAD综合征(发病急骤肥胖与下丘脑功能障碍,通气不足和自主神经失调) - 一,其中一人开始快速增加体重和贪食症,呼吸道疾病患极为罕见的疾病,暂停呼吸睡眠,肺泡通气不足,以及心肺功能停止时。此外,在患者的诊断特性缺乏应对二氧化碳水平的增加的。
今天,在世界上注册的约100情况下,这种障碍。它通常出现在10(一般为3年)的年龄,显然有一种遗传性的。尽管研究在西方,该综合征ROHHAD的病因尚不清楚进行。据认为,它产生于垂体功能障碍,这导致遗传突变。然而,科学家们还没有确定哪个进程被违反在这种情况下。
作者:纳塔利娅Kienya
资料来源:T&P I>
BLOCKQUOTE>
通过factroom.ru