Жизнь — интересная!
Подписывайтесь на нашу группу в Telegram и Facebook, чтобы быть в сообществе единомышленников, находить вдохновение и не пропускать свежие и удивительные статьи с bashny.net.
886
0.2
2015-09-08
5 самых невообразимых генетических патологий человека
Бывает же такое….
Фатальная семейная бессонница, детская старость и лишние кости в неположенных местах — это не только идеи, вдохновляющие кинорежиссеров, но и реальные наследственные болезни, которые возникают в результате генетических мутаций. «Теории и практики» составили список самых невообразимых заболеваний и описали причины их появления.
Фатальная семейная бессонница
Фатальная семейная бессонница — редчайшая наследственная болезнь, при которой человек погибает от неспособности заснуть. До сих пор она отмечалась лишь в сорока семьях по всему миру. Фатальная бессонница обычно проявляется между 30 и 60 годами (чаще всего — после 50 лет) и продолжается от 7 до 36 месяцев. По мере того как заболевание прогрессирует, пациент страдает от все более тяжелых нарушений сна, причем никакие снотворные ему не помогают. На первой стадии бессонница сопровождается паническими атаками и фобиями, на второй к ним прибавляются галлюцинации и повышенное потоотделение. На третьей стадии болезни человек полностью теряет способность спать и начинает выглядеть намного старше своих лет. Затем развивается деменция, и пациент погибает — как правило, от истощения или пневмонии.
Смертельная бессонница возникает из-за того, что в кодоне (кодирующем тринуклеотиде) 178 гена PRNP, расположенного в 20-й хромосоме, вместо аспарагина, который в норме присутствует в организме в составе белков и необходим для нормальной работы нервной системы, появляется аспарагиновая кислота. Это меняет форму белковой молекулы, и она превращается в прион — агрессивный аномальный белок, в составе которого нет нуклеиновых кислот. Под действием одного приона окружающие молекулы уподобляются ему, и это ведет к необратимым изменениям. Прежде чего, они протекают в тканях таламуса: подкорковой «станции» всех видов чувствительности, отвечающей в том числе за возникновение сонного состояния и двигательные функции: глотание, сосание, жевание, смех. Под действием прионов ядра таламуса покрываются порами, превращаются в губку и перестают работать.
Для болезни характерен аутосомно-доминантный тип наследования: то есть у нее нет носителей. Детям она передается от родителей с вероятностью 50% и только при условии, если кто-то из них болен. Мужчины и женщины болеют фатальной семейной бессонницей с одинаковой частотой. На сегодняшний день это заболевание считается неизлечимым.
Нарколепсия-катаплексия
Синдром нарколепсии-катаплексии, для которого характерны внезапные приступы сна и расслабления мускулатуры тела, тоже имеет генетическую природу и возникает из-за нарушений быстрой фазы сна. Он встречается намного чаще фатальной семейной бессонницы: у сорока из каждых ста тысяч человек, в равной степени у мужчин и у женщин. Человек, страдающий нарколепсией, способен внезапно заснуть на несколько минут посреди дня. «Сонные атаки» напоминают фазу быстрого сна и могут случаться очень часто — до ста раз в день, с предшествующей им головной болью либо без нее. Они часто провоцируются бездеятельностью, но могут возникать в совершенно неподходящее время: во время полового акта, занятий спортом, вождения. Просыпается человек отдохнувшим.
Примерно в 80% случаев нарколепсии сопутствует катаплексия: внезапная эпизодическая потеря мышечного тонуса, которая повторяется регулярно. В легких случаях у пациента слегка отвисает нижняя челюсть и возникает ощущении слабости в коленях, но если состояние тяжелое, человек может внезапно упасть на ровном месте. Его сознание при этом остается ясным. Катаплексия развивается на фоне выраженных эмоциональных реакций: смеха, злости, страха или удивления, что делает подобное состояние особенно неудобным.
Причина возникновения болезни пока ясна не до конца, но в ряде случаев отмечалась мутация нейромедиатора орексина (ген HCRT, 17q21), который регулирует процесс передачи возбуждающих сигналов в мозгу и влияет на сон и аппетит. Сигнальная система между орексинергическими и другими нейронами дает сбой, угнетается активность моноаминергических нейронов и снижается приток возбуждающих сигналов в кору.
Устранить нарколепсию-катаплексию невозможно, однако симптоматическое лечение есть. Пациенты начинают чувствовать себя лучше благодаря регулярному сну в строго определенное время и препаратам, активизирующим работу центральных адренергических систем.
Фибродисплазия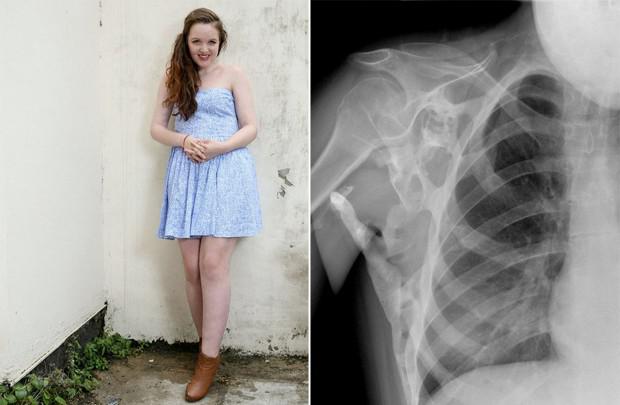
Фибродисплазия оссифицирующая прогрессирующая (ФОП) — редкое генетическое заболевание, при котором организм начинает формировать новые кости — оссификаты — в неположенных местах: внутри мышц, связок, сухожилий и других соединительных тканей. К их образованию может привести любая травма: ушиб, порез, перелом, внутримышечная инъекция или операция. Из-за этого удалять оссификаты нельзя: после хирургического вмешательства кость может только сильнее разрастись. Физиологически оссификаты не отличаются от обыкновенных костей и могут выдерживать значительные нагрузки, вот только находятся не там, где надо.
ФОП возникает из-за мутации в гене ACVR1/ALK2, кодирующем рецептор костного морфогенетического белка. Она передается человеку по наследству от одного из родителей, если он тоже болен. Быть носителем этого заболевания нельзя: пациент либо болен, либо нет. Пока ФОП относится к числу неизлечимых болезней, однако сейчас проводится вторая серия испытаний препарата под названием паловаротен, который позволяет заблокировать ген, ответственный за патологию.
Прогерия
Детская прогерия, или синдром Хатчинсона-Гилфорда, — заболевание, которым страдает один человек из четырех-семи миллионов. Организм детей с таким диагнозом стареет чрезвычайно быстро и рано: уже в подростковом возрасте пациенты выглядят и чувствуют себя, как старики. У них развивается множество старческих патологий, возникают нарушения в работе внутренних органов и систем, а кости, кожа, мышцы и сухожилия становятся слабыми и вялыми. При этом по уровню развития дети с прогерией не уступают сверстникам, а иногда и опережают их. Средняя продолжительность жизни людей, страдающих синдромом Хатчинсона-Гилфорда, — 13 лет. Как правило, причиной смерти становится инфаркт миокарда. Описан всего один случай, когда пациент с таким диагнозом дожил до 45 лет.
Причина возникновения детской прогерии — спонтанная мутация в одном из двух копий (аллелей) гена LMNA, кодирующего преламин, А, который является предшественником зрелых форм ламинов, А и С. Эти белки необходимы для того, чтобы оболочка клеточных ядер была целой и нормально функционировала. Мутантный преламин, А — или «прогерин», как предлагают называть его некоторые авторы, — накапливается в клетках, так что их ядерные оболочки сморщиваются, а ядра приобретают неправильную форму. Такие клетки уже не могут нормально делиться, так что организм теряет способность не только расти, но и заменять отмирающие клетки новыми.
На данный момент это заболевание также считается неизлечимым, однако продлить жизнь пациентов и улучшить ее качество помогает разнообразное симптоматическое лечение и физическая активность, особенно плавание. Эти средства позволяют улучшить состояние кровеносной системы и суставов. Кроме того, используется гормон роста.
Синдром РОХХАД
Синдромом РОХХАД (Rapid-onset Obesity with Hypothalamic dysfunction, Hypoventilation and Autonomic Dysregulation) — чрезвычайно редкое заболевание, при котором человек начинает стремительно набирать вес и страдает от булимии, респираторных болезней, остановок дыхания во сне, альвеолярной гиповентиляции и кардиореспираторных остановок. Также для пациентов с таким диагнозом характерно отсутствие реакции на повышение в крови углекислого газа.
На сегодняшний день в мире зарегистрировано порядка 100 случаев возникновения этого расстройства. Обычно оно проявляется в возрасте до 10 лет (чаще всего около 3 лет) и, по всей видимости, имеет наследственную природу. Несмотря на проведенные на Западе исследования, этиология синдрома РОХХАД до сих пор не ясна. Считается, что он появляется из-за дисфункции гипофиза, которую вызывает генетическая мутация. Однако ученым еще только предстоит определить, какой именно процесс нарушается в этом случае.
via factroom.ru

Фатальная семейная бессонница, детская старость и лишние кости в неположенных местах — это не только идеи, вдохновляющие кинорежиссеров, но и реальные наследственные болезни, которые возникают в результате генетических мутаций. «Теории и практики» составили список самых невообразимых заболеваний и описали причины их появления.
Фатальная семейная бессонница

Фатальная семейная бессонница — редчайшая наследственная болезнь, при которой человек погибает от неспособности заснуть. До сих пор она отмечалась лишь в сорока семьях по всему миру. Фатальная бессонница обычно проявляется между 30 и 60 годами (чаще всего — после 50 лет) и продолжается от 7 до 36 месяцев. По мере того как заболевание прогрессирует, пациент страдает от все более тяжелых нарушений сна, причем никакие снотворные ему не помогают. На первой стадии бессонница сопровождается паническими атаками и фобиями, на второй к ним прибавляются галлюцинации и повышенное потоотделение. На третьей стадии болезни человек полностью теряет способность спать и начинает выглядеть намного старше своих лет. Затем развивается деменция, и пациент погибает — как правило, от истощения или пневмонии.
Смертельная бессонница возникает из-за того, что в кодоне (кодирующем тринуклеотиде) 178 гена PRNP, расположенного в 20-й хромосоме, вместо аспарагина, который в норме присутствует в организме в составе белков и необходим для нормальной работы нервной системы, появляется аспарагиновая кислота. Это меняет форму белковой молекулы, и она превращается в прион — агрессивный аномальный белок, в составе которого нет нуклеиновых кислот. Под действием одного приона окружающие молекулы уподобляются ему, и это ведет к необратимым изменениям. Прежде чего, они протекают в тканях таламуса: подкорковой «станции» всех видов чувствительности, отвечающей в том числе за возникновение сонного состояния и двигательные функции: глотание, сосание, жевание, смех. Под действием прионов ядра таламуса покрываются порами, превращаются в губку и перестают работать.
Для болезни характерен аутосомно-доминантный тип наследования: то есть у нее нет носителей. Детям она передается от родителей с вероятностью 50% и только при условии, если кто-то из них болен. Мужчины и женщины болеют фатальной семейной бессонницей с одинаковой частотой. На сегодняшний день это заболевание считается неизлечимым.
Нарколепсия-катаплексия

Синдром нарколепсии-катаплексии, для которого характерны внезапные приступы сна и расслабления мускулатуры тела, тоже имеет генетическую природу и возникает из-за нарушений быстрой фазы сна. Он встречается намного чаще фатальной семейной бессонницы: у сорока из каждых ста тысяч человек, в равной степени у мужчин и у женщин. Человек, страдающий нарколепсией, способен внезапно заснуть на несколько минут посреди дня. «Сонные атаки» напоминают фазу быстрого сна и могут случаться очень часто — до ста раз в день, с предшествующей им головной болью либо без нее. Они часто провоцируются бездеятельностью, но могут возникать в совершенно неподходящее время: во время полового акта, занятий спортом, вождения. Просыпается человек отдохнувшим.
Примерно в 80% случаев нарколепсии сопутствует катаплексия: внезапная эпизодическая потеря мышечного тонуса, которая повторяется регулярно. В легких случаях у пациента слегка отвисает нижняя челюсть и возникает ощущении слабости в коленях, но если состояние тяжелое, человек может внезапно упасть на ровном месте. Его сознание при этом остается ясным. Катаплексия развивается на фоне выраженных эмоциональных реакций: смеха, злости, страха или удивления, что делает подобное состояние особенно неудобным.
Причина возникновения болезни пока ясна не до конца, но в ряде случаев отмечалась мутация нейромедиатора орексина (ген HCRT, 17q21), который регулирует процесс передачи возбуждающих сигналов в мозгу и влияет на сон и аппетит. Сигнальная система между орексинергическими и другими нейронами дает сбой, угнетается активность моноаминергических нейронов и снижается приток возбуждающих сигналов в кору.
Устранить нарколепсию-катаплексию невозможно, однако симптоматическое лечение есть. Пациенты начинают чувствовать себя лучше благодаря регулярному сну в строго определенное время и препаратам, активизирующим работу центральных адренергических систем.
Фибродисплазия

Фибродисплазия оссифицирующая прогрессирующая (ФОП) — редкое генетическое заболевание, при котором организм начинает формировать новые кости — оссификаты — в неположенных местах: внутри мышц, связок, сухожилий и других соединительных тканей. К их образованию может привести любая травма: ушиб, порез, перелом, внутримышечная инъекция или операция. Из-за этого удалять оссификаты нельзя: после хирургического вмешательства кость может только сильнее разрастись. Физиологически оссификаты не отличаются от обыкновенных костей и могут выдерживать значительные нагрузки, вот только находятся не там, где надо.
ФОП возникает из-за мутации в гене ACVR1/ALK2, кодирующем рецептор костного морфогенетического белка. Она передается человеку по наследству от одного из родителей, если он тоже болен. Быть носителем этого заболевания нельзя: пациент либо болен, либо нет. Пока ФОП относится к числу неизлечимых болезней, однако сейчас проводится вторая серия испытаний препарата под названием паловаротен, который позволяет заблокировать ген, ответственный за патологию.
Прогерия

Детская прогерия, или синдром Хатчинсона-Гилфорда, — заболевание, которым страдает один человек из четырех-семи миллионов. Организм детей с таким диагнозом стареет чрезвычайно быстро и рано: уже в подростковом возрасте пациенты выглядят и чувствуют себя, как старики. У них развивается множество старческих патологий, возникают нарушения в работе внутренних органов и систем, а кости, кожа, мышцы и сухожилия становятся слабыми и вялыми. При этом по уровню развития дети с прогерией не уступают сверстникам, а иногда и опережают их. Средняя продолжительность жизни людей, страдающих синдромом Хатчинсона-Гилфорда, — 13 лет. Как правило, причиной смерти становится инфаркт миокарда. Описан всего один случай, когда пациент с таким диагнозом дожил до 45 лет.
Причина возникновения детской прогерии — спонтанная мутация в одном из двух копий (аллелей) гена LMNA, кодирующего преламин, А, который является предшественником зрелых форм ламинов, А и С. Эти белки необходимы для того, чтобы оболочка клеточных ядер была целой и нормально функционировала. Мутантный преламин, А — или «прогерин», как предлагают называть его некоторые авторы, — накапливается в клетках, так что их ядерные оболочки сморщиваются, а ядра приобретают неправильную форму. Такие клетки уже не могут нормально делиться, так что организм теряет способность не только расти, но и заменять отмирающие клетки новыми.
На данный момент это заболевание также считается неизлечимым, однако продлить жизнь пациентов и улучшить ее качество помогает разнообразное симптоматическое лечение и физическая активность, особенно плавание. Эти средства позволяют улучшить состояние кровеносной системы и суставов. Кроме того, используется гормон роста.
Синдром РОХХАД

Синдромом РОХХАД (Rapid-onset Obesity with Hypothalamic dysfunction, Hypoventilation and Autonomic Dysregulation) — чрезвычайно редкое заболевание, при котором человек начинает стремительно набирать вес и страдает от булимии, респираторных болезней, остановок дыхания во сне, альвеолярной гиповентиляции и кардиореспираторных остановок. Также для пациентов с таким диагнозом характерно отсутствие реакции на повышение в крови углекислого газа.
На сегодняшний день в мире зарегистрировано порядка 100 случаев возникновения этого расстройства. Обычно оно проявляется в возрасте до 10 лет (чаще всего около 3 лет) и, по всей видимости, имеет наследственную природу. Несмотря на проведенные на Западе исследования, этиология синдрома РОХХАД до сих пор не ясна. Считается, что он появляется из-за дисфункции гипофиза, которую вызывает генетическая мутация. Однако ученым еще только предстоит определить, какой именно процесс нарушается в этом случае.
Автор: Наталия Киеня
Источник: T&P
via factroom.ru
Портал БАШНЯ. Копирование, Перепечатка возможна при указании активной ссылки на данную страницу.