Жизнь — интересная!
Подписывайтесь на нашу группу в Telegram и Facebook, чтобы быть в сообществе единомышленников, находить вдохновение и не пропускать свежие и удивительные статьи с bashny.net.
686
0.1
2016-09-29
Настоящая чёрная смерть
Её по праву называют «королевой опухолей». Она меняет свои маски, словно на маскараде, вводя в заблуждение даже самых опытных специалистов. Убивает исподтишка, подобно дворцовым интриганам, “отравляя” организм человека в кратчайшие сроки. Перед её могуществом трепещет мировое онкологическое сообщество. Она научит ценить аристократическую бледность.
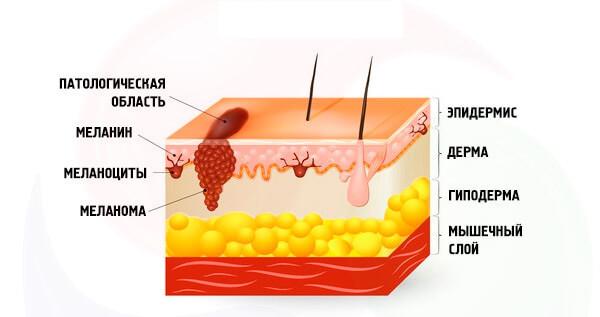
Меланома кожи является результатом неопластической трансформации меланоцитов – клеток, продуцирующих различные вариации пигмента меланина. Меланоциты произрастают из нервного гребня – группы клеток маргинальной части нервного желоба.
Клетки нервного гребня отличаются высоким уровнем миграции, и большинство из них направляется вниз от нервной трубки, образуя разнообразные нервные и эндокринные структуры. Однако меланобласты проявляют свою уникальность ещё до потенциального приобретения опухолевого фенотипа и мигрируют не вниз, а в стороны от нервной трубки.
Молекулярные механизмы данного процесса по-прежнему остаются неизвестными. Судьбу меланобласта сложно назвать предопределённой: данный элемент впоследствии может развиться в нейрон, лейомиоцит, а в случае высокого уровня меланоцит-глиального потенциала – и в компонент глии.
После дифференцировки в меланоците выделяют тело (сому) и отростки, которые располагаются в базальном и шиповатом слоях эпидермиса соответственно. Под действием меланоцитстимулирующих и адренокортикотропного гормонов, а также солнечного света в меланосомах синтезируется (эу)меланин (и феомеланин), функцией которого является защита ядерного аппарата клетки от повреждения УФ-излучением.
Синтезированный меланин транспортируется в шиповатый слой эпидермиса по отросткам меланоцита, далее в кератиноциты эпидермиса, придавая коже загар. Спустя некоторое время данный полимер гидролизируется в лизосомах, а коже возвращается её привычный оттенок.
Данный вариант развития событий можно назвать идеальным. Но что же происходит у тех, кому повезло меньше? Ключевым моментом инициации неопластической трансформации меланоцита является нелетальная мутация в его генетическом аппарате. Причиной такого повреждения может быть самое разное воздействие: как физическое, так и химическое.
Как и у большинства опухолей, причины запуска онкогенеза при меланоме можно объединить по нескольким признакам:
1) Подавление активности генов-онкосупрессоров;
2) Активация протоонкогенов;
3) Экзогенные мутации, полученные самыми разнообразными путями.
Что же делает меланому столь опасной? Казалось бы, наружная локализация, всем известные алгоритмы на этапе самообследования, высокая иммуногенность опухоли должны сделать её терапию максимально эффективной.
Однако непредсказуемость метастатического каскада вкупе с отсутствием специально разработанной терапии метастатических меланом, непредсказуемость клинической и морфологической – как микро-, так и макроскопической – форм и высочайшая частота рецидивирования приводят к тому, что на сегодняшний день заболеваемость меланомой увеличивается во всех странах мира; медиана выживаемости при меланоме составляет не более 8-ми месяцев, а эффективность радикального хирургического лечения не превышает 65%. Ответить на вопросы клиницистов и пациентов могут лишь учёные.
Возможность секвенирования генома позволила углубиться в понимание канцерогенеза при меланоме кожи. Изучение процессов-инициаторов при трансформации меланоцитов в самую агрессивную из ныне существующих опухолей является самым важным этапом в изучении не только меланомы кожи, но и всех остальных опухолей.
Только путём определения новых мишеней возможны назначение таргетной терапии, расширение диагностических горизонтов, а также возможен пересмотр классификаций по клинико-морфологическим формам с предпочтением объединения подвидов меланом исходя из характера мутаций. С учётом этого, на обзор механизмов канцерогенеза при меланоме нужно обратить особое внимание.
Одним из наиболее важных достижений в изучении канцерогенеза при меланоме по праву считается анализ механизмов сигнальной трансдукции, которые активируются в процессе её развития. Исходя из этиологического фактора, обнаружение тех или иных мутаций в меланоме кожи может ответить на вопрос взаимосвязи этиологии и патогенеза, клинических форм и терапевтических решений.
Что такое сигнальный путь? Это некая «эстафета», при которой в строго определенном порядке задействуется последовательность тех или иных молекул, которые участвуют в передаче любой разновидности сигнала от клеточного рецептора внутрь клетки. Сигнальный путь может быть активирован экзогенно – факторами роста, нейромедиаторами, гормонами – или эндогенно – при помощи сигналов-стартеров, активируемых по разным причинам внутри клетки.
Эпидемиология
Средний возраст заболевших меланомой кожи составляет примерно 45 лет, однако за последние годы меланома стала всё чаще возникать у совсем молодых людей (15-25 лет). Эта опухоль встречается у мужчин и женщин, причём у женщин в 1,5-2 раза чаще. По статистике, на каждые 100.000 здоровых человек приходится 14 больных с меланомой.
В 2006 г. в России зарегистрировано 7.364 новых больных меланомой кожи и 3.033 смертей. Ежегодное число заболевших меланомой в России составляет 5.700 человек, 2.200 из которых, вероятнее всего, погибнут от данного заболевания.
В различных регионах мира показатели заболеваемости меланомой существенно отличаются. По данным ВОЗ, за период 1988–2012 гг. наиболее высокие стандартизованные показатели заболеваемости меланомой кожи 25–29,8о /оооо были характерны для белого населения Австралии и Новой Зеландии. Достаточно высокий уровень заболеваемости 15–18,6о /оооо отмечен среди европейцев, живущих в Зимбабве, белых мужчин США (Лос-Анжелес, Сан-Франциско), женщин Австрии, Норвегии.
Высокий, по меркам Европы, уровень в 8,8–14,1о /оооо был среди жителей Дании, Италии, Швейцарии, Швеции, мужчин Австрии и Норвегии. Самые низкие стандартизованные показатели заболеваемости меланомой кожи 0,1–1,5о /оооо выявлены в Алжире, у индейцев и черных жителей США, Уганды, Зимбабве, в Китае, Корее, Японии.
На возникновение меланомы кожи влияет и расовая принадлежность человека. Согласно статистике ВОЗ, чернокожие в 4 раза реже страдают от меланомы, чем европеоиды. Существует предположение, что более тёмная кожа содержит большее количество меланина в эпидермисе, что способствует более сильной задержке УФ-излучения, и тем самым является более надёжной защитой от его повреждающего действия на меланоциты, выработанной эволюционно в соответствии с климатическими условиями у проживающих в данных областях земного шара людей.
Соответственно, народы со слабой естественной защитой (меньшим количеством меланина в коже) чаще страдают меланомой. Так, например, среди жителей Казахстана было отмечено восьмикратное повышение заболеваемости меланомой у приезжих с белой кожей по сравнению с представителями коренного населения.
Таким образом, данные статистики ВОЗ позволяют удостовериться, что чем выше природное количество меланоцитов в коже, тем ниже риск возникновения меланомы, следовательно, низкий уровень пигментации является фактором риска.
Оценка данного фактора возможна уже при обычном клиническом осмотре. Люди, которые относятся к “светлому” фенотипу, обычно слабо восприимчивы к солнечному загару и очень предрасположены к развитию солнечных ожогов из-за высокой чувствительности к УФ-излучению, благодаря чему риск развития меланомы возрастает у них в два раза.
Другим признаком нарушения уровня пигментации является наличие большого количества веснушек на коже, которые обусловлены скоплением пролиферирующих меланоцитов в базальном слое эпидермиса и эпителии наружных отделов волосяных фолликулов.
Крайне тяжелая степень нарушения пигментации, характеризующаяся полным отсутствием меланина в организме, наблюдается у альбиносов. Канцерогенное действие УФ-излучения у них настолько велико, что у большинства к 20 годам возникают предопухолевые поражения или опухоли кожи, и настолько губительно, что менее 10% этих людей доживают до 30 лет.
Сигнальные пути
На сегодняшний день наиболее изученными являются следующие сигнальные пути:
Сложности начинаются у учёных при попытке отследить строгую специфичность сигнального пути и регулируемого им процесса. Ошибочно полагать, будто активация этих сигнальных путей является признаком неопластической трансформации в меланоме.
Во-первых, данные сигнальные пути существуют и в норме, регулируя дифференцировку клеток в антенатальном и постнатальном периодах, обеспечивают воспроизведение эффектов тех или иных гормонов и прочие типовые процессы, направленные на поддержание нормальной жизнедеятельности клетки.
Во-вторых, данные пути задействованы не только в меланоцитах: они являются универсальными сигнальными системами, задействованными в большинстве клеток организма человека. В чём же дело? В любом из сигнальных путей может произойти та или иная генетическая или эпигенетическая поломка, результатом которой станет негативная регуляция сигнального пути с исходом в канцерогенез.
MAPK
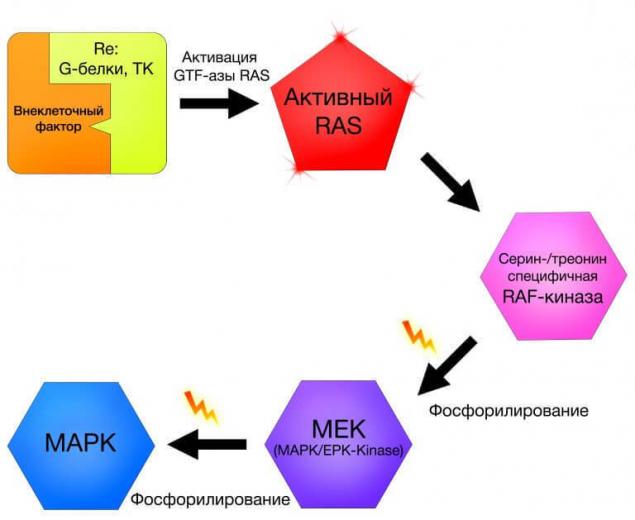
К кластеру МАРК (mitogen-activated protein kinase) относятся внутриклеточные сигнальные пути самых разнообразных функций [8]. Критерием для включения в МАРК-группу является наличие митоген-активируемых протеинкиназ в модуле, в котором, помимо киназ, содержатся протеинфосфатазы и белки-сборщики белковых вспомогательных комплексов [9].
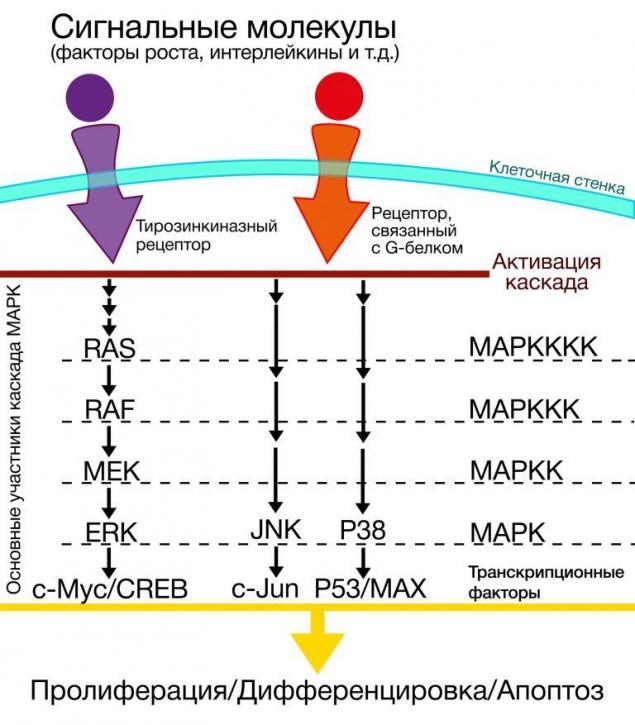
Сами по себе ни МАРК-кластер, ни его эффекты не представляют особого интереса в контексте канцерогенеза, однако те мутации, которые возникают в генах белков, участвующих в сигнальных путях в клетках меланомы кожи, требуют более детального рассмотрения. Среди множества киназ, задействованных при передаче сигнала, в кластере МАРК существуют так называемые Raf-киназы.
Raf- – это семейство серин-треонин зависимых протеинкиназ, название которых является акронимом от Rapidly Accelerated Fibrosarcoma. Но где быстрорастущая фибросаркома, а где меланома кожи?
Дело в том, что в глубинных механизмах онкогенеза нередко обнаруживаются перекресты путей, и такие, как в нашем случае, в том числе. В контексте данной статьи не имеет никакого смысла останавливаться на подробном представлении этапов онкогенеза фибросарком, поэтому остановимся особенностях активности Raf- при меланоме кожи.
BRAF
Примечательным в семействе Raf- для нас может быть B-raf – белок, играющий роль в МАРК-каскаде, чьими эффектами являются дифференцировка и деление клеток. Белок B-raf кодируется одноименным геном, который был бы абсолютно не примечателен, если бы не одно «но»: возникающая под действием чрезмерного УФ-облучения в данном гене V600[x] мутация (под [x] в данном случае подразумевается обозначение несинонимичной замены, приводящей к мутации), заключающаяся в замене валина на лейцин (V600L), лизин (V600K) или глутаминовую кислоту (V600E) в 600-ой позиции, служит стартером неопластической трансформации в меланоме кожи, а также является мишенью для действия лекарственных средств, объединенных в группу ингибиторов BRAF V600L. Это так называемые «-нибы»: иматиниб, сорафениб, вемурафениб и т.п.
В рандомизированных клинических исследованиях была обнаружена интересная особенность: если при верифицированном мутированном BRAF-статусе пациента низкомолекулярные ингибиторы BRAF приводили к торможению развития меланомы, то потребление «-нибов» пациентами, мутации BRAF у которых вызывали сомнения либо отсутствовали, приводило к мутации другого каскада — RAS-RAF-MEK-ERK, патологически активируя его и инициируя неопластическую трансформацию.
Таким образом, можно сделать вывод, что низкомолекулярные BRAF-ингибиторы хоть и являются эффективным терапевтическим средством, всё же демонстрируют опасную амбивалентность в случае недостаточной диагностики или ошибочной интерпретации BRAF-статуса пациента.
C 2011 г. альтернативой химиотерапии для пациентов с метастатической меланомой кожи с мутациями BRAF V600E является препарат вемурафениб (зелораф). В основе его действия лежит ингибирование димеризации BRAF, поскольку при мутации V600E происходит усиление сигналинга ERK в результате димеризации мутантной киназы.
В I фазе испытаний ответ на вемурафениб был получен у 81 % больных меланомой с мутацией V600E (уменьшение в диаметре всех опухолевых узлов на 30 %). В III фазе испытаний общая выживаемость в течение 6 мес отмечена у 84 % пациентов с метастатической меланомой, выживаемость без прогрессирования составила 5–7 мес.
Однако у 20–30 % пациентов, получающих вемурафениб, наблюдаются побочные эффекты: малигнизация доброкачественных поражений и появление плоскоклеточного рака кожи, что связывают с активацией пути MAPK через мутации RAS при подавлении RAF. У некоторых пациентов при лечении возникают новые очаги меланомы с диким типом BRAF. Недавние исследования показывают, что МАРК поддается более эффективному ингибированию при использовании не только BRAF (в режиме монотерапии), но и МЕК-ингибиторов.
Данная комбинация не только более эффективна, она предотвращает развитие резистентности к проводимой терапии, а также возникновение наиболее серьёзного нежелательного эффекта BRAF-ингибиторов – плоскоклеточного рака кожи.
Предполагают, что эффект может иметь комбинация препаратов сорафениба и лонафарниба – ингибитора фарнезилирования (реакции, приводящей к инициации канцерогенной активности белка RAS) и «заякоривания» RAS на мембране клетки.
В январе 2014 г. зарегистрирован новый таргетный препарат дабрафениб для лечения меланомы с мутацией в 600-м кодоне BRAF. В отличие от вемурафениба дабрафениб действует не только при замене V600E, но и при мутации V600K. Оба ингибитора BRAF были первыми препаратами, которые имели эффект у пациентов с метастазами в мозг. К сожалению, практически у всех пациентов, ответивших на вемурафениб, с течением времени появляется устойчивость к терапии.
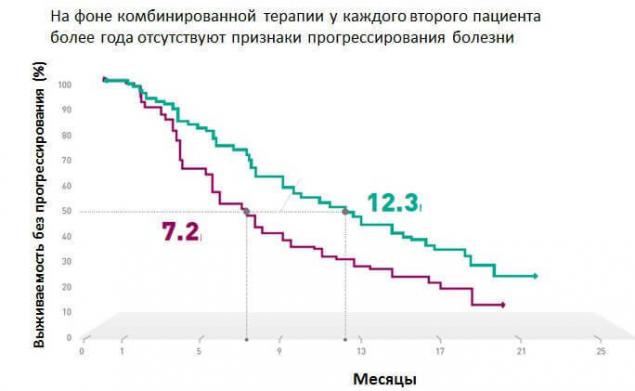
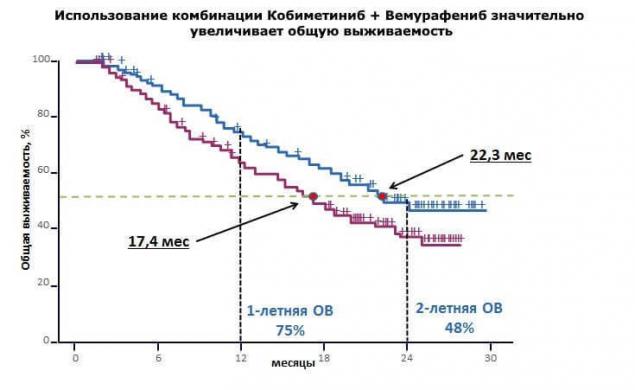
«В рамках клинического исследования coBRIM было подтверждено, что комбинированная терапия с использованием препаратов вемурафениб и кобиметиниб в 90% случаев позволяет достичь ответа на терапию у больных BRAF- положительной метастатической меланомой. У каждого второго пациента более года отсутствуют признаки прогрессирования заболевания, а общая выживаемость приближается к двум годам».
МЕК
МЕК является серин-треониновой киназой МАРК-кластера, активируемой в связи с изменениями в вышестоящих звеньях сигнальной цепи.
Роль МЕК заключается в интеграции сигналов от различных факторов роста с последующей активацией пролиферации. Являясь частью МАРК-каскада, МЕК участвует в регуляции активности транскрипционных факторов, таких как, например, C-myc.
Белок Myc, кодируемый одноимённым геном и являющийся протоонкогеном, является не только «каноничным» примером транскрипторного фактора, но и контролирует структуру хроматина, регулируя ацетилирование гистонов, что в свою очередь влияет на активность экспрессии генов. Мутантный ген Mус, находящийся в 8-ой хромосоме, обнаруживается во многих видах опухолей; классическим примером являетсятранслокация t (8;14), приводящая к возникновению и развитию лимфомы Бёркитта.
Эксперименты, направленные на его ингибирование, приводили к лизису опухолевых клеток лёгкого у мышей. Следовательно, Мус является терапевтической мишенью, в частности, разработка селективных аллостерических ингибиторов Мус является актуальной темой для клинических исследований.
NRAS
N-RAS (Neuroblastoma-Ras) – ген, кодирующий одноименный белок, входящий в так называемое суперсемейство Ras – малых ГТФ-аз. Наряду с NRAS в данное семейство входят гены K-Ras, H-Ras (вызывающие неопластическую трансформацию при заражении вирусом саркомы Кристен и Харви соответственно) и др. На сегодняшний день функция NRAS определена как передача пролиферативных сигналов от рецепторов факторов роста. NRAS, как и все суперсемейство, задействованы в сигнальном пути МАРК.
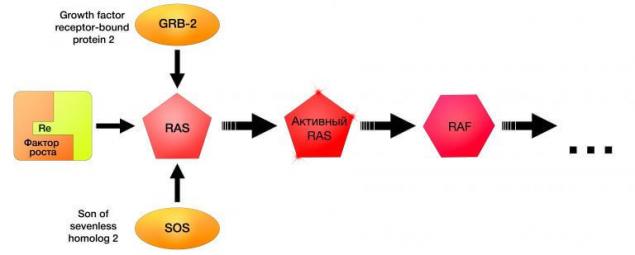
Мутации NRAS обнаруживались в 10-26% случаев меланомы кожи, преимущественно облучаемой УФ. Ингибирование NRAS открыло новые горизонты в терапии меланом, показав на практике значение тесной взаимосвязи всех звеньев сигнального пути. Первым препаратом таргетной терапии меланомы с мутацией NRAS является ингибитор MEK162, благодаря приему которого практически у 70% больных данной категории удается получить реакцию на лечение и добиться стабилизации заболевания.
c-KIT
Последним рассматриваемым в контексте данной статьи звеном МАРК-каскада станет ген c-KIT. Последним, но не по значению: этот ген, являющийся протоонкогеном, белок которого является рецептором факторов роста стволовых клеток, выявляется в каждом третьем случае меланомы различных локализаций: как кожи, так и слизистых.
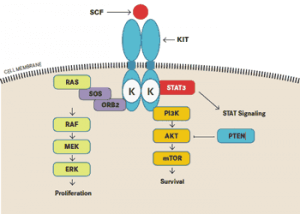
Схема сигнального пути Kit
Как и для многих других мутаций, существуют лекарственные средства, направленные на ингибирование мутаций Kit. Учитывая непрерываемую взаимодополняющую связь звеньев сигнального пути МАРК, не вызывает удивления тот факт, что Kit-мутация успешно ингибируется уже известными по таргетной терапии BRAF-положительных меланом «-нибами»: Иматинибом, Нилатинибом и другими лекарственными средствами.
PI3K – фосфоинозитол-3-киназный путь
Следующим компонентом пролиферативных сигнальных путей, патологически активированных в клетках меланомы, является PI3K – фосфоинозитол-3-киназный путь, нередко обозначаемый в литературе как «сигнальный путь PI3K/AKT/mTOR». Это каскад реакций, основные события которого разворачиваются вокруг следующих ферментов: фосфоинозитид-3-киназы (PI3K), семейства протеинкиназ В, компонентами которого являются серин-треонин киназы АКТ 1, 2, 3, и ещё одной серин-треонинспецифичной киназы, получившей название «мишень рапамицина» и обозначаемой как mTOR.
Данный сигнальный путь является универсальным для большинства клеток человеческого организма и обеспечивает пролиферацию клеток, метаболизм и дифференцировку.
В контексте канцерогенеза в клетках меланомы кожи наибольший интерес представляют компоненты AKT и PTEN.
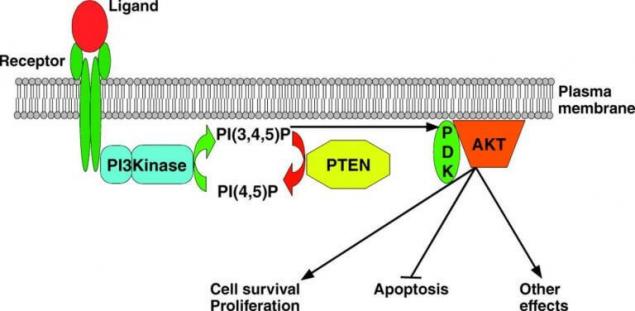
Классический путь PTEN
PTEN
PTEN представляет собой фермент фосфатазу, являющуюся негативным регулятором РI3K-пути и кодируемую одноименным геном-онкосупрессором. Путём отщепления фосфатной группы у фосфатидилинозитол-3-фосфатов данный фермент тормозит (вплоть до полной блокады) проведение сигнала по фосфоинозитол-3-киназному пути. Инактивация PTEN обнаруживается во многих опухолях, поскольку приводит к неконтролируемому делению с утратой дифференцировки, сбоям в метаболизме клетки и извращённому синтезу. Инактивирующие PTEN мутации обнаруживаются в 10-30% меланом.
АКТ
Другим компонентом PI3K-сигнального пути, рассматриваемым в вопросе развития меланом, станет семейство АКТ.
Семейство АКТ представляет собой группу киназ, которые путём присоединения остатков фосфорной кислоты к различным белкам цитозоля контролируют их активность. Данное семейство выполняет как классические функции, например, регуляцию пролиферации, дифференцировку и изменение цитоскелета, так и щекотливые в контексте канцерогенеза функции ангионеогенеза, ухода от апоптоза и приобретения резистентности к цитостатикам .
В семейство АКТ входит 3 подвида протеинкиназ: АКТ-1, АКТ-2 и АКТ-3, являющихся продуктами соответствующих генов.
Akt1 — α-серин/треониновая протеинкиназа – ингибирование апоптоза, биосинтез белка (в сторону «плюс-ткани», гипертрофия миоцитов и т.п.).
Akt2 — ß -серин/треониновая протеинкиназа- участвует в метаболизме инсулина, индуцирует транспорт глюкозы, осуществляемый ГЛЮТ-4.
Akt3 — γ- серин/треониновой-протеинкиназы – функция достоверно неизвестна.
Различные виды АКТ гиперэкспрессированы в 45-70% меланом. Эксперименты, проведённые с фосфатазой PHLPP, являющейся катализатором дефосфорилирования в молекуле АКТ-1 с последующей инактивацией АКТ-1, привели к апоптозу и замедлению пролиферации в неопластически измененных клетках.
mTOR
Данная протеинкиназа также показывает треонин-сериновую специфичность, существующую в виде двух внутриклеточных комплексов: TORC1 и TORC2. Мишенью рапамицина называется первый комплекс, являющийся терапевтической мишенью одноименного иммуносупрессора.
Но mammalian Тarget Of Rapamycin интересует нас сейчас не как мишень рапамицина, а как компонент PI3K-сигнального пути, играющий ключевую роль в процессе клеточного роста. Несмотря на то, что молекула TOR является неделимым производным двух её субъединиц, последние обладают совершенно разными полномочиями.
Повреждение первого комплекса не несёт каких-либо катастрофических последствий для клетки, а нарушение TOR2 приведёт к так называемому «аресту» (прекращению) клеточного цикла на этапе фазы G2/M фазе через несколько поколений. Если пострадают обе субъединицы, то клетка «замрёт» в фазе G0 в следующем поколении. Таким образом, становится понятно: самостоятельно ли, совместно ли с ТОR1, но так или иначе ТОR2 оказывает сильнейшее влияние на клеточный цикл.
Следующим рассматриваемым в данной статье компонентом системы контроля клеточного цикла является система циклинзасимых киназ CDKN2A, CDK4 и CDK6. Мутации, делеции, гиперметилирование промотора являются нарушениями, обнаруживаемыми в 50% (CDKN2A) и 10-12% (CDK4) случаев меланомы, а наследуемые герминальные мутации в генах предрасположенности могут обусловливать возникновение семейной меланомы в 15% случаев.
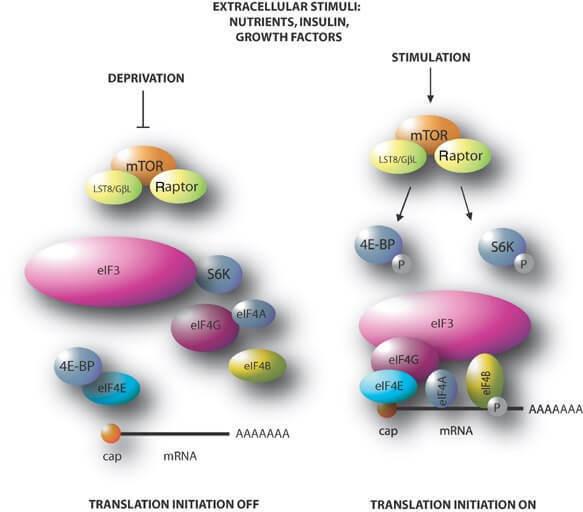
Рисунок 1 Механизм действия мишени рапамицина
CDKN2A (cyclin-dependent kinase Inhibitor 2A) – ген, локализованный в коротком плече 9 хромосомы и кодирующий два белка, которые являются онкосупрессорами: р14 и р16. При помощи данных белков регулируется активность, пожалуй, самого известного онкосупрессора р53 и белка ретинобластомы.
В случае мутации CDKN2A (дупликация кодона R112) оба продукта данного гена подвергаются поломке. Что интересно, данная мутация является доминантной в случаях наследственной меланомы в семьях скандинавских национальностей. Мутации данного гена обнаружены в 30-40% случаев меланом, особенно часто они встречаются в контексте семейной меланомы. Генный полиморфизм имеет значение, определяя скорость развития опухоли и её резистентность к полихимиотерапии.
Однако ошибочно полагать, что CDKN2A ответственен исключительно за семейные случаи заболевания. Спорадические соматические мутации CDKN2A встречаются в 50% случаев меланомы кожи, реализуемой либо путём деактивации р16, либо путём метилирования промотора (10%).
Белковый комплекс циклина-D и CDK4 регулирует экспрессию белка ретинобластомы, влияющего на активность клеточного цикла и пролиферацию клеток. Приведенный ниже каскад реакций схематично показывает последовательность событий в генетическом аппарате и самой клетке при поломке более высоких механизмов регуляции, в данном случае – белка р16:
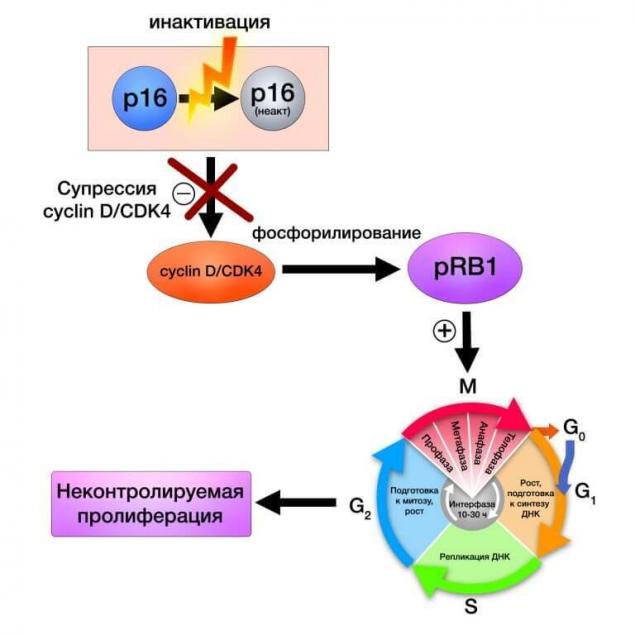
CDK4 (сyclin-dependent kinase 4) – фермент, кодируемый одноимённым геном и являющийся частью семьи циклинзависимых киназ. Будучи звеном единой системы контроля клеточного цикла, CDK4, как и CDK6, связан с соответствующими протеинами и белком ретинобластомы.
В контексте канцерогенеза данные элементы выступают в роли рычагов управления клеточным циклом, неконтролируемая активность которого может быть результатом тех или иных нарушений в управляющих ферментах. CDK как молекулярная мишень интересует не только дерматоонкологов, поскольку ингибиторы CDK как целый класс лекарственных противоопухолевых препаратов широко используется для лечения самых разнообразных видов онкологических заболеваний, таких как эстроген-положительный и HER-отрицательный рак молочной железы, лейкемия, рак щитовидной железы (палбоциклиб, летрозол, абемациклиб).
Ингибиторы циклинзависимой киназы блокируют активность соответствующего фермента как самостоятельно, так в комплексе «фермент+циклин»; действуют данные препараты, как правило, в фазе G1 клеточного цикла. Снижение активности CDK может быть обеспечено, например, снижением уровня экспрессии генов циклинов или повышением степени распада циклинов.
Ингибиторы CDK не только обеспечивают адекватный рост клетки, но и обладают способностью остановки цикла на стадии G1 в ответ на повреждение ДНК, неблагоприятные внешние условия, etc.
WNT
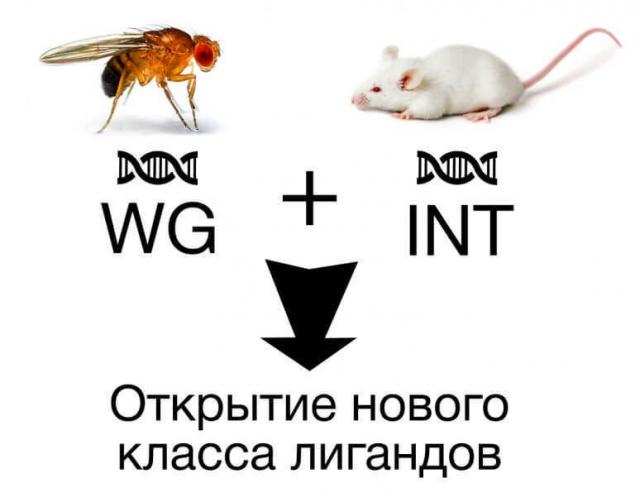
Каскад WNT является примером классического сигнального пути, регулируя пролиферативную активность и дифференцировку клеток. Свое название данный путь получил в ходе слияния названий двух генов: WG мухи-дрозофилы, который подавлял рост крыльев, и INT, который изначально рассматривался в ходе исследований рака молочной железы у мышей. В единую структуру белок был объединен исходя из ряда функций, который позволил выделить новый класс лигандов.
По своей структуре WNT представляет сложный комплекс, при графическом изображении которого можно заметить сходство с раскрытой ладонью. Стоит отметить, что уровень гликозилирования никак не влияет на секреторную активность данного комплекса, однако N-гликозилирование повышает секрецию WNT.
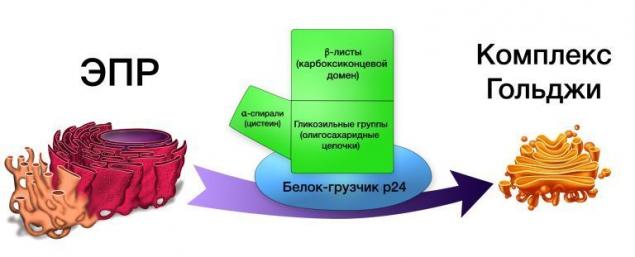
Белок WNT экскретируется белками комплекса Гольджи, такими как, например, GPR177, и переносится на поверхность клетки с помощью «белков-грузчиков» p24.
Действовать на клетку WNT может согласно каноническому или неканоническому пути, они же — β-катенин зависимый и β-катенин независимый пути соответственно. В первом случае изменяется концентрация клеточного β-катенина, конечным звеном чего становится контроль генетической экспрессии, влияющей на морфогенез клетки, а во втором случае регулируется полярность клетки путём стимуляции реорганизации цитоскелета и изменения интенсивности кальциевого обмена.
Каноничный путь представляет собой последовательность реакций, направленных на накопление ß-катенина в клетке и непосредственную его доставку в ядро, где тот будет действовать как транскрипционный фактор.
Путь WNT начинается с cоединения WNT с рецептором к липопротеинам низкой плотности (Low-density lipoprotein Relater Protein-5, LRP-5), далее путём активации влияния белка AXIN1 АРС (негативный регулятор концентрации ß-катенина, мутации гена которого приводят к возникновению и развитию колоректального рака), киназ GSK3, CK1 (киназа гликоге
Источник: medach.pro/surgery/onkologiya-hirurgicheskie-distsiplinyi/melanoma/
Меланома кожи является результатом неопластической трансформации меланоцитов – клеток, продуцирующих различные вариации пигмента меланина. Меланоциты произрастают из нервного гребня – группы клеток маргинальной части нервного желоба.

Клетки нервного гребня отличаются высоким уровнем миграции, и большинство из них направляется вниз от нервной трубки, образуя разнообразные нервные и эндокринные структуры. Однако меланобласты проявляют свою уникальность ещё до потенциального приобретения опухолевого фенотипа и мигрируют не вниз, а в стороны от нервной трубки.
Молекулярные механизмы данного процесса по-прежнему остаются неизвестными. Судьбу меланобласта сложно назвать предопределённой: данный элемент впоследствии может развиться в нейрон, лейомиоцит, а в случае высокого уровня меланоцит-глиального потенциала – и в компонент глии.
После дифференцировки в меланоците выделяют тело (сому) и отростки, которые располагаются в базальном и шиповатом слоях эпидермиса соответственно. Под действием меланоцитстимулирующих и адренокортикотропного гормонов, а также солнечного света в меланосомах синтезируется (эу)меланин (и феомеланин), функцией которого является защита ядерного аппарата клетки от повреждения УФ-излучением.
Синтезированный меланин транспортируется в шиповатый слой эпидермиса по отросткам меланоцита, далее в кератиноциты эпидермиса, придавая коже загар. Спустя некоторое время данный полимер гидролизируется в лизосомах, а коже возвращается её привычный оттенок.
Данный вариант развития событий можно назвать идеальным. Но что же происходит у тех, кому повезло меньше? Ключевым моментом инициации неопластической трансформации меланоцита является нелетальная мутация в его генетическом аппарате. Причиной такого повреждения может быть самое разное воздействие: как физическое, так и химическое.
Как и у большинства опухолей, причины запуска онкогенеза при меланоме можно объединить по нескольким признакам:
1) Подавление активности генов-онкосупрессоров;
2) Активация протоонкогенов;
3) Экзогенные мутации, полученные самыми разнообразными путями.
Что же делает меланому столь опасной? Казалось бы, наружная локализация, всем известные алгоритмы на этапе самообследования, высокая иммуногенность опухоли должны сделать её терапию максимально эффективной.
Однако непредсказуемость метастатического каскада вкупе с отсутствием специально разработанной терапии метастатических меланом, непредсказуемость клинической и морфологической – как микро-, так и макроскопической – форм и высочайшая частота рецидивирования приводят к тому, что на сегодняшний день заболеваемость меланомой увеличивается во всех странах мира; медиана выживаемости при меланоме составляет не более 8-ми месяцев, а эффективность радикального хирургического лечения не превышает 65%. Ответить на вопросы клиницистов и пациентов могут лишь учёные.
Возможность секвенирования генома позволила углубиться в понимание канцерогенеза при меланоме кожи. Изучение процессов-инициаторов при трансформации меланоцитов в самую агрессивную из ныне существующих опухолей является самым важным этапом в изучении не только меланомы кожи, но и всех остальных опухолей.
Только путём определения новых мишеней возможны назначение таргетной терапии, расширение диагностических горизонтов, а также возможен пересмотр классификаций по клинико-морфологическим формам с предпочтением объединения подвидов меланом исходя из характера мутаций. С учётом этого, на обзор механизмов канцерогенеза при меланоме нужно обратить особое внимание.
Одним из наиболее важных достижений в изучении канцерогенеза при меланоме по праву считается анализ механизмов сигнальной трансдукции, которые активируются в процессе её развития. Исходя из этиологического фактора, обнаружение тех или иных мутаций в меланоме кожи может ответить на вопрос взаимосвязи этиологии и патогенеза, клинических форм и терапевтических решений.
Что такое сигнальный путь? Это некая «эстафета», при которой в строго определенном порядке задействуется последовательность тех или иных молекул, которые участвуют в передаче любой разновидности сигнала от клеточного рецептора внутрь клетки. Сигнальный путь может быть активирован экзогенно – факторами роста, нейромедиаторами, гормонами – или эндогенно – при помощи сигналов-стартеров, активируемых по разным причинам внутри клетки.
Эпидемиология
Средний возраст заболевших меланомой кожи составляет примерно 45 лет, однако за последние годы меланома стала всё чаще возникать у совсем молодых людей (15-25 лет). Эта опухоль встречается у мужчин и женщин, причём у женщин в 1,5-2 раза чаще. По статистике, на каждые 100.000 здоровых человек приходится 14 больных с меланомой.
В 2006 г. в России зарегистрировано 7.364 новых больных меланомой кожи и 3.033 смертей. Ежегодное число заболевших меланомой в России составляет 5.700 человек, 2.200 из которых, вероятнее всего, погибнут от данного заболевания.
В различных регионах мира показатели заболеваемости меланомой существенно отличаются. По данным ВОЗ, за период 1988–2012 гг. наиболее высокие стандартизованные показатели заболеваемости меланомой кожи 25–29,8о /оооо были характерны для белого населения Австралии и Новой Зеландии. Достаточно высокий уровень заболеваемости 15–18,6о /оооо отмечен среди европейцев, живущих в Зимбабве, белых мужчин США (Лос-Анжелес, Сан-Франциско), женщин Австрии, Норвегии.
Высокий, по меркам Европы, уровень в 8,8–14,1о /оооо был среди жителей Дании, Италии, Швейцарии, Швеции, мужчин Австрии и Норвегии. Самые низкие стандартизованные показатели заболеваемости меланомой кожи 0,1–1,5о /оооо выявлены в Алжире, у индейцев и черных жителей США, Уганды, Зимбабве, в Китае, Корее, Японии.
На возникновение меланомы кожи влияет и расовая принадлежность человека. Согласно статистике ВОЗ, чернокожие в 4 раза реже страдают от меланомы, чем европеоиды. Существует предположение, что более тёмная кожа содержит большее количество меланина в эпидермисе, что способствует более сильной задержке УФ-излучения, и тем самым является более надёжной защитой от его повреждающего действия на меланоциты, выработанной эволюционно в соответствии с климатическими условиями у проживающих в данных областях земного шара людей.
Соответственно, народы со слабой естественной защитой (меньшим количеством меланина в коже) чаще страдают меланомой. Так, например, среди жителей Казахстана было отмечено восьмикратное повышение заболеваемости меланомой у приезжих с белой кожей по сравнению с представителями коренного населения.
Таким образом, данные статистики ВОЗ позволяют удостовериться, что чем выше природное количество меланоцитов в коже, тем ниже риск возникновения меланомы, следовательно, низкий уровень пигментации является фактором риска.
Оценка данного фактора возможна уже при обычном клиническом осмотре. Люди, которые относятся к “светлому” фенотипу, обычно слабо восприимчивы к солнечному загару и очень предрасположены к развитию солнечных ожогов из-за высокой чувствительности к УФ-излучению, благодаря чему риск развития меланомы возрастает у них в два раза.
Другим признаком нарушения уровня пигментации является наличие большого количества веснушек на коже, которые обусловлены скоплением пролиферирующих меланоцитов в базальном слое эпидермиса и эпителии наружных отделов волосяных фолликулов.
Крайне тяжелая степень нарушения пигментации, характеризующаяся полным отсутствием меланина в организме, наблюдается у альбиносов. Канцерогенное действие УФ-излучения у них настолько велико, что у большинства к 20 годам возникают предопухолевые поражения или опухоли кожи, и настолько губительно, что менее 10% этих людей доживают до 30 лет.
Сигнальные пути
На сегодняшний день наиболее изученными являются следующие сигнальные пути:
- МАРК
- PI3K
- CDK
- Wnt
Сложности начинаются у учёных при попытке отследить строгую специфичность сигнального пути и регулируемого им процесса. Ошибочно полагать, будто активация этих сигнальных путей является признаком неопластической трансформации в меланоме.
Во-первых, данные сигнальные пути существуют и в норме, регулируя дифференцировку клеток в антенатальном и постнатальном периодах, обеспечивают воспроизведение эффектов тех или иных гормонов и прочие типовые процессы, направленные на поддержание нормальной жизнедеятельности клетки.
Во-вторых, данные пути задействованы не только в меланоцитах: они являются универсальными сигнальными системами, задействованными в большинстве клеток организма человека. В чём же дело? В любом из сигнальных путей может произойти та или иная генетическая или эпигенетическая поломка, результатом которой станет негативная регуляция сигнального пути с исходом в канцерогенез.
MAPK
К кластеру МАРК (mitogen-activated protein kinase) относятся внутриклеточные сигнальные пути самых разнообразных функций [8]. Критерием для включения в МАРК-группу является наличие митоген-активируемых протеинкиназ в модуле, в котором, помимо киназ, содержатся протеинфосфатазы и белки-сборщики белковых вспомогательных комплексов [9].

Сами по себе ни МАРК-кластер, ни его эффекты не представляют особого интереса в контексте канцерогенеза, однако те мутации, которые возникают в генах белков, участвующих в сигнальных путях в клетках меланомы кожи, требуют более детального рассмотрения. Среди множества киназ, задействованных при передаче сигнала, в кластере МАРК существуют так называемые Raf-киназы.
Raf- – это семейство серин-треонин зависимых протеинкиназ, название которых является акронимом от Rapidly Accelerated Fibrosarcoma. Но где быстрорастущая фибросаркома, а где меланома кожи?
Дело в том, что в глубинных механизмах онкогенеза нередко обнаруживаются перекресты путей, и такие, как в нашем случае, в том числе. В контексте данной статьи не имеет никакого смысла останавливаться на подробном представлении этапов онкогенеза фибросарком, поэтому остановимся особенностях активности Raf- при меланоме кожи.
BRAF
Примечательным в семействе Raf- для нас может быть B-raf – белок, играющий роль в МАРК-каскаде, чьими эффектами являются дифференцировка и деление клеток. Белок B-raf кодируется одноименным геном, который был бы абсолютно не примечателен, если бы не одно «но»: возникающая под действием чрезмерного УФ-облучения в данном гене V600[x] мутация (под [x] в данном случае подразумевается обозначение несинонимичной замены, приводящей к мутации), заключающаяся в замене валина на лейцин (V600L), лизин (V600K) или глутаминовую кислоту (V600E) в 600-ой позиции, служит стартером неопластической трансформации в меланоме кожи, а также является мишенью для действия лекарственных средств, объединенных в группу ингибиторов BRAF V600L. Это так называемые «-нибы»: иматиниб, сорафениб, вемурафениб и т.п.
В рандомизированных клинических исследованиях была обнаружена интересная особенность: если при верифицированном мутированном BRAF-статусе пациента низкомолекулярные ингибиторы BRAF приводили к торможению развития меланомы, то потребление «-нибов» пациентами, мутации BRAF у которых вызывали сомнения либо отсутствовали, приводило к мутации другого каскада — RAS-RAF-MEK-ERK, патологически активируя его и инициируя неопластическую трансформацию.
Таким образом, можно сделать вывод, что низкомолекулярные BRAF-ингибиторы хоть и являются эффективным терапевтическим средством, всё же демонстрируют опасную амбивалентность в случае недостаточной диагностики или ошибочной интерпретации BRAF-статуса пациента.
C 2011 г. альтернативой химиотерапии для пациентов с метастатической меланомой кожи с мутациями BRAF V600E является препарат вемурафениб (зелораф). В основе его действия лежит ингибирование димеризации BRAF, поскольку при мутации V600E происходит усиление сигналинга ERK в результате димеризации мутантной киназы.
В I фазе испытаний ответ на вемурафениб был получен у 81 % больных меланомой с мутацией V600E (уменьшение в диаметре всех опухолевых узлов на 30 %). В III фазе испытаний общая выживаемость в течение 6 мес отмечена у 84 % пациентов с метастатической меланомой, выживаемость без прогрессирования составила 5–7 мес.

Однако у 20–30 % пациентов, получающих вемурафениб, наблюдаются побочные эффекты: малигнизация доброкачественных поражений и появление плоскоклеточного рака кожи, что связывают с активацией пути MAPK через мутации RAS при подавлении RAF. У некоторых пациентов при лечении возникают новые очаги меланомы с диким типом BRAF. Недавние исследования показывают, что МАРК поддается более эффективному ингибированию при использовании не только BRAF (в режиме монотерапии), но и МЕК-ингибиторов.
Данная комбинация не только более эффективна, она предотвращает развитие резистентности к проводимой терапии, а также возникновение наиболее серьёзного нежелательного эффекта BRAF-ингибиторов – плоскоклеточного рака кожи.
Предполагают, что эффект может иметь комбинация препаратов сорафениба и лонафарниба – ингибитора фарнезилирования (реакции, приводящей к инициации канцерогенной активности белка RAS) и «заякоривания» RAS на мембране клетки.
В январе 2014 г. зарегистрирован новый таргетный препарат дабрафениб для лечения меланомы с мутацией в 600-м кодоне BRAF. В отличие от вемурафениба дабрафениб действует не только при замене V600E, но и при мутации V600K. Оба ингибитора BRAF были первыми препаратами, которые имели эффект у пациентов с метастазами в мозг. К сожалению, практически у всех пациентов, ответивших на вемурафениб, с течением времени появляется устойчивость к терапии.

«В рамках клинического исследования coBRIM было подтверждено, что комбинированная терапия с использованием препаратов вемурафениб и кобиметиниб в 90% случаев позволяет достичь ответа на терапию у больных BRAF- положительной метастатической меланомой. У каждого второго пациента более года отсутствуют признаки прогрессирования заболевания, а общая выживаемость приближается к двум годам».

МЕК
МЕК является серин-треониновой киназой МАРК-кластера, активируемой в связи с изменениями в вышестоящих звеньях сигнальной цепи.
Роль МЕК заключается в интеграции сигналов от различных факторов роста с последующей активацией пролиферации. Являясь частью МАРК-каскада, МЕК участвует в регуляции активности транскрипционных факторов, таких как, например, C-myc.

Белок Myc, кодируемый одноимённым геном и являющийся протоонкогеном, является не только «каноничным» примером транскрипторного фактора, но и контролирует структуру хроматина, регулируя ацетилирование гистонов, что в свою очередь влияет на активность экспрессии генов. Мутантный ген Mус, находящийся в 8-ой хромосоме, обнаруживается во многих видах опухолей; классическим примером являетсятранслокация t (8;14), приводящая к возникновению и развитию лимфомы Бёркитта.
Эксперименты, направленные на его ингибирование, приводили к лизису опухолевых клеток лёгкого у мышей. Следовательно, Мус является терапевтической мишенью, в частности, разработка селективных аллостерических ингибиторов Мус является актуальной темой для клинических исследований.
NRAS
N-RAS (Neuroblastoma-Ras) – ген, кодирующий одноименный белок, входящий в так называемое суперсемейство Ras – малых ГТФ-аз. Наряду с NRAS в данное семейство входят гены K-Ras, H-Ras (вызывающие неопластическую трансформацию при заражении вирусом саркомы Кристен и Харви соответственно) и др. На сегодняшний день функция NRAS определена как передача пролиферативных сигналов от рецепторов факторов роста. NRAS, как и все суперсемейство, задействованы в сигнальном пути МАРК.

Мутации NRAS обнаруживались в 10-26% случаев меланомы кожи, преимущественно облучаемой УФ. Ингибирование NRAS открыло новые горизонты в терапии меланом, показав на практике значение тесной взаимосвязи всех звеньев сигнального пути. Первым препаратом таргетной терапии меланомы с мутацией NRAS является ингибитор MEK162, благодаря приему которого практически у 70% больных данной категории удается получить реакцию на лечение и добиться стабилизации заболевания.
c-KIT
Последним рассматриваемым в контексте данной статьи звеном МАРК-каскада станет ген c-KIT. Последним, но не по значению: этот ген, являющийся протоонкогеном, белок которого является рецептором факторов роста стволовых клеток, выявляется в каждом третьем случае меланомы различных локализаций: как кожи, так и слизистых.

Схема сигнального пути Kit
Как и для многих других мутаций, существуют лекарственные средства, направленные на ингибирование мутаций Kit. Учитывая непрерываемую взаимодополняющую связь звеньев сигнального пути МАРК, не вызывает удивления тот факт, что Kit-мутация успешно ингибируется уже известными по таргетной терапии BRAF-положительных меланом «-нибами»: Иматинибом, Нилатинибом и другими лекарственными средствами.
PI3K – фосфоинозитол-3-киназный путь
Следующим компонентом пролиферативных сигнальных путей, патологически активированных в клетках меланомы, является PI3K – фосфоинозитол-3-киназный путь, нередко обозначаемый в литературе как «сигнальный путь PI3K/AKT/mTOR». Это каскад реакций, основные события которого разворачиваются вокруг следующих ферментов: фосфоинозитид-3-киназы (PI3K), семейства протеинкиназ В, компонентами которого являются серин-треонин киназы АКТ 1, 2, 3, и ещё одной серин-треонинспецифичной киназы, получившей название «мишень рапамицина» и обозначаемой как mTOR.
Данный сигнальный путь является универсальным для большинства клеток человеческого организма и обеспечивает пролиферацию клеток, метаболизм и дифференцировку.
В контексте канцерогенеза в клетках меланомы кожи наибольший интерес представляют компоненты AKT и PTEN.

Классический путь PTEN
PTEN
PTEN представляет собой фермент фосфатазу, являющуюся негативным регулятором РI3K-пути и кодируемую одноименным геном-онкосупрессором. Путём отщепления фосфатной группы у фосфатидилинозитол-3-фосфатов данный фермент тормозит (вплоть до полной блокады) проведение сигнала по фосфоинозитол-3-киназному пути. Инактивация PTEN обнаруживается во многих опухолях, поскольку приводит к неконтролируемому делению с утратой дифференцировки, сбоям в метаболизме клетки и извращённому синтезу. Инактивирующие PTEN мутации обнаруживаются в 10-30% меланом.
АКТ
Другим компонентом PI3K-сигнального пути, рассматриваемым в вопросе развития меланом, станет семейство АКТ.
Семейство АКТ представляет собой группу киназ, которые путём присоединения остатков фосфорной кислоты к различным белкам цитозоля контролируют их активность. Данное семейство выполняет как классические функции, например, регуляцию пролиферации, дифференцировку и изменение цитоскелета, так и щекотливые в контексте канцерогенеза функции ангионеогенеза, ухода от апоптоза и приобретения резистентности к цитостатикам .
В семейство АКТ входит 3 подвида протеинкиназ: АКТ-1, АКТ-2 и АКТ-3, являющихся продуктами соответствующих генов.
Akt1 — α-серин/треониновая протеинкиназа – ингибирование апоптоза, биосинтез белка (в сторону «плюс-ткани», гипертрофия миоцитов и т.п.).
Akt2 — ß -серин/треониновая протеинкиназа- участвует в метаболизме инсулина, индуцирует транспорт глюкозы, осуществляемый ГЛЮТ-4.
Akt3 — γ- серин/треониновой-протеинкиназы – функция достоверно неизвестна.
Различные виды АКТ гиперэкспрессированы в 45-70% меланом. Эксперименты, проведённые с фосфатазой PHLPP, являющейся катализатором дефосфорилирования в молекуле АКТ-1 с последующей инактивацией АКТ-1, привели к апоптозу и замедлению пролиферации в неопластически измененных клетках.
mTOR
Данная протеинкиназа также показывает треонин-сериновую специфичность, существующую в виде двух внутриклеточных комплексов: TORC1 и TORC2. Мишенью рапамицина называется первый комплекс, являющийся терапевтической мишенью одноименного иммуносупрессора.
Но mammalian Тarget Of Rapamycin интересует нас сейчас не как мишень рапамицина, а как компонент PI3K-сигнального пути, играющий ключевую роль в процессе клеточного роста. Несмотря на то, что молекула TOR является неделимым производным двух её субъединиц, последние обладают совершенно разными полномочиями.
Повреждение первого комплекса не несёт каких-либо катастрофических последствий для клетки, а нарушение TOR2 приведёт к так называемому «аресту» (прекращению) клеточного цикла на этапе фазы G2/M фазе через несколько поколений. Если пострадают обе субъединицы, то клетка «замрёт» в фазе G0 в следующем поколении. Таким образом, становится понятно: самостоятельно ли, совместно ли с ТОR1, но так или иначе ТОR2 оказывает сильнейшее влияние на клеточный цикл.
Следующим рассматриваемым в данной статье компонентом системы контроля клеточного цикла является система циклинзасимых киназ CDKN2A, CDK4 и CDK6. Мутации, делеции, гиперметилирование промотора являются нарушениями, обнаруживаемыми в 50% (CDKN2A) и 10-12% (CDK4) случаев меланомы, а наследуемые герминальные мутации в генах предрасположенности могут обусловливать возникновение семейной меланомы в 15% случаев.

Рисунок 1 Механизм действия мишени рапамицина
CDKN2A (cyclin-dependent kinase Inhibitor 2A) – ген, локализованный в коротком плече 9 хромосомы и кодирующий два белка, которые являются онкосупрессорами: р14 и р16. При помощи данных белков регулируется активность, пожалуй, самого известного онкосупрессора р53 и белка ретинобластомы.
В случае мутации CDKN2A (дупликация кодона R112) оба продукта данного гена подвергаются поломке. Что интересно, данная мутация является доминантной в случаях наследственной меланомы в семьях скандинавских национальностей. Мутации данного гена обнаружены в 30-40% случаев меланом, особенно часто они встречаются в контексте семейной меланомы. Генный полиморфизм имеет значение, определяя скорость развития опухоли и её резистентность к полихимиотерапии.
Однако ошибочно полагать, что CDKN2A ответственен исключительно за семейные случаи заболевания. Спорадические соматические мутации CDKN2A встречаются в 50% случаев меланомы кожи, реализуемой либо путём деактивации р16, либо путём метилирования промотора (10%).
Белковый комплекс циклина-D и CDK4 регулирует экспрессию белка ретинобластомы, влияющего на активность клеточного цикла и пролиферацию клеток. Приведенный ниже каскад реакций схематично показывает последовательность событий в генетическом аппарате и самой клетке при поломке более высоких механизмов регуляции, в данном случае – белка р16:

CDK4 (сyclin-dependent kinase 4) – фермент, кодируемый одноимённым геном и являющийся частью семьи циклинзависимых киназ. Будучи звеном единой системы контроля клеточного цикла, CDK4, как и CDK6, связан с соответствующими протеинами и белком ретинобластомы.
В контексте канцерогенеза данные элементы выступают в роли рычагов управления клеточным циклом, неконтролируемая активность которого может быть результатом тех или иных нарушений в управляющих ферментах. CDK как молекулярная мишень интересует не только дерматоонкологов, поскольку ингибиторы CDK как целый класс лекарственных противоопухолевых препаратов широко используется для лечения самых разнообразных видов онкологических заболеваний, таких как эстроген-положительный и HER-отрицательный рак молочной железы, лейкемия, рак щитовидной железы (палбоциклиб, летрозол, абемациклиб).
Ингибиторы циклинзависимой киназы блокируют активность соответствующего фермента как самостоятельно, так в комплексе «фермент+циклин»; действуют данные препараты, как правило, в фазе G1 клеточного цикла. Снижение активности CDK может быть обеспечено, например, снижением уровня экспрессии генов циклинов или повышением степени распада циклинов.
Ингибиторы CDK не только обеспечивают адекватный рост клетки, но и обладают способностью остановки цикла на стадии G1 в ответ на повреждение ДНК, неблагоприятные внешние условия, etc.
WNT
Каскад WNT является примером классического сигнального пути, регулируя пролиферативную активность и дифференцировку клеток. Свое название данный путь получил в ходе слияния названий двух генов: WG мухи-дрозофилы, который подавлял рост крыльев, и INT, который изначально рассматривался в ходе исследований рака молочной железы у мышей. В единую структуру белок был объединен исходя из ряда функций, который позволил выделить новый класс лигандов.

По своей структуре WNT представляет сложный комплекс, при графическом изображении которого можно заметить сходство с раскрытой ладонью. Стоит отметить, что уровень гликозилирования никак не влияет на секреторную активность данного комплекса, однако N-гликозилирование повышает секрецию WNT.

Белок WNT экскретируется белками комплекса Гольджи, такими как, например, GPR177, и переносится на поверхность клетки с помощью «белков-грузчиков» p24.
Действовать на клетку WNT может согласно каноническому или неканоническому пути, они же — β-катенин зависимый и β-катенин независимый пути соответственно. В первом случае изменяется концентрация клеточного β-катенина, конечным звеном чего становится контроль генетической экспрессии, влияющей на морфогенез клетки, а во втором случае регулируется полярность клетки путём стимуляции реорганизации цитоскелета и изменения интенсивности кальциевого обмена.
Каноничный путь представляет собой последовательность реакций, направленных на накопление ß-катенина в клетке и непосредственную его доставку в ядро, где тот будет действовать как транскрипционный фактор.
Путь WNT начинается с cоединения WNT с рецептором к липопротеинам низкой плотности (Low-density lipoprotein Relater Protein-5, LRP-5), далее путём активации влияния белка AXIN1 АРС (негативный регулятор концентрации ß-катенина, мутации гена которого приводят к возникновению и развитию колоректального рака), киназ GSK3, CK1 (киназа гликоге
Источник: medach.pro/surgery/onkologiya-hirurgicheskie-distsiplinyi/melanoma/
Портал БАШНЯ. Копирование, Перепечатка возможна при указании активной ссылки на данную страницу.