686
Справжня чорна смерть.
«Королева пам’яті». Вона змінює маски, як якщо на маскараді, вводить в оману навіть найдосвідченіших фахівців. Вбиває кришку, як палацові інтригуари, «поісонування» тіла людини в найкоротші терміни. До своєї влади глобальні збудники раку. Ви навчите Вас оцінити аристократичну блідість.
Маланома шкіри є результатом неопластичної трансформації меланоцитів – клітин, які виробляють різні варіації пігментного меланіну. Меланоцити виростають з нервового гребеня - група клітин маргінальної частини нервової кори.
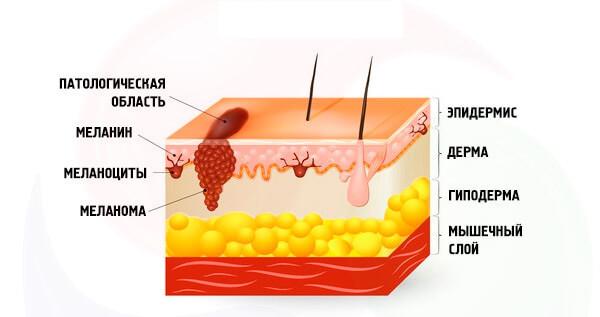
Нервові клітини мають високий рівень міграції, а більшість з них спрямовуються з нервової труби, що утворюють різні нервові та ендокринні структури. Тим не менш, меланобласти показують свою унікальність навіть перед потенційним придбанням пухлинного фенотипу і мігрувати не внизу вгору, але далеко від нервової труби.
Молекулярні механізми цього процесу досі невідомі. Доля меланоблапласту навряд чи може бути викликана заздалегідь: цей елемент може згодом розвиватися в нейрон, лейоміоцит, а в разі високого рівня меланоцито-гліального потенціалу - і в компоненту глі.
Після диференціації в меланоциті, тіло (сома) і процеси виділяються, які розташовані в базальних і шипкових шарах епідермісу, відповідно. Під дією меланоцимуляційних і адренакортикотропних гормонів, а також сонячних променів, меаносом синтезу (ев)меланін (і пекомланін), функція якого полягає в тому, щоб захистити ядерний апарат клітин від пошкодження ультрафіолетового випромінювання.
Сингабаритний меланінов перевозиться в спінний шар епідермісу уздовж процесів меланоциту, потім в кератиноцити епідермісу, що дає шкірі тан. Після деякого часу цей полімер гідролізується в лізосом, і шкіра повертає свій звичний відтінок.
Цей сценарій можна назвати ідеальним. Але що відбувається для тих, хто менш завадити? Ключ до ініціювання неопластичної трансформації меланоциту – це нелетична мутація в її генетичному апараті. Причиною такого пошкодження може бути різні ефекти: як фізичні, так і хімічні.
Як при більшості новоутворень причини онкогенезу в меланомі можна поєднувати за кількома ознаками:
1) Пригнічення активності генів онкосупресора;
2) Активація прото-oncogenes;
(3) Екзогенні мутації, отримані в різних напрямках.
Що таке меланома? Здавалося б, що зовнішня локалізація, відомі алгоритми на стадії самовизначення, висока імуногенність пухлини повинна максимально ефективно проводити її терапія.
Однак непередбачуваність метастатичного каскаду, пов'язаної з відсутністю спеціально розробленої терапії метастатичної меланоми, непередбачуваності клінічних і морфологічних форм - як мікро-, так і макроскопічної - і найвищою частотою рецидиву призводять до того, що сьогодні захворюваність меланоми зростає в усіх країнах світу; рівень медіанності меланоми не більше 8 місяців, а ефективність радикального хірургічного лікування не перевищує 65%. Відповіді можна відповісти тільки клініки та пацієнти.
Можливість занурення геному дозволили нам проникнути в розуміння карциногенезу в меланомі шкіри. Дослідження процесів, які ініціюють трансформацію меланоцитів в найбільш агресивні існуючі пухлини, є найважливішим кроком у дослідженні не тільки меланоми шкіри, але й інших пухлин.
Тільки шляхом виявлення нових цілей можна призначати цільову терапію, розширити діагностичні горизонти, а також переглянути класифікацію за допомогою клініко-морфологічних форм з перевагами поєднання підвидів меланоми на основі природи мутацій. З цим на увазі слід звертати особливу увагу огляд механізмів карциногенезу в меланомі.
Одним з найважливіших досягнень у дослідженні карциногенезу в меланомі є аналіз механізмів передачі сигналів, які активуються під час його розвитку. На основі етіологічного фактора виявлення певних мутацій в меланомі шкіри може відповісти питання взаємозв’язків етіології та патогенезу, клінічних форм і терапевтичних рішень.
Що таке сигнал? Це свого роду «релей», в якому в строго визначеному порядку бере участь послідовність певних молекул, які беруть участь у передачі будь-якого виду сигналу від рецептора клітин в клітинку. Шлях сигналізації може бути активований екзогенно—розривні чинники, нейротрансмітери, гормони — або ендогенно—за сигналами стартера, активовані з різних причин в клітині.
епідеміологія
Середній вік пацієнтів меланоми становить близько 45 років, але в останні роки меланома стала все більш поширеною в дуже молодих людей (15-25 років). Ця пухлина зустрічається у чоловіків і жінок, і у жінок 1,5-2 рази частіше. За статистикою, для кожного 100 000 здорових людей, є 14 пацієнтів з меланоми.
У 2006 році в Росії зареєстровано 7364 нових хворих на меланоми шкіри та 3,033 смерті. Річна кількість випадків меланоми в Росії становить 5,700 осіб, 2,200 з яких, ймовірно, відмирають від захворювання.
У різних регіонах світу значно змінюється захворюваність меланоми. Згідно з ВООЗ, між 1988 та 2012 року, найвищий стандартизований захворюваність меланоми шкіри (25–29.8o/ooh) серед білих популяцій Австралії та Нової Зеландії. Серед європейців, які живуть в Зімбабве, білі чоловіки США (Лес-Анджелес, Сан-Франциско), жінки Австрії, Норвегії.
Високий рівень 8,8-14.1 o /ooh був серед жителів Данії, Італії, Швейцарії, Швеції, чоловіків Австрії та Норвегії. Найнижчий стандартизований захворюванні меланоми шкіри 0,1-1,5 ° /ooh виявлений в Алжирі, в індійських і чорних жителів США, Уганда, Зімбабве, Китай, Корея, Японія.
Поява меланоми шкіри також впливає на расу людини. Згідно з статистикою ВООЗ, чорношкірі в 4 рази рідше страждають від меланоми, ніж кавказький. Існує припущення, що темніше шкіра містить більше меланіну в епідермісі, що сприяє більшій затримці в УФ-випромінюванні, і таким чином є більш надійним захистом від шкідливого ефекту на меланоцитах, розробленої еволюціонально відповідно до кліматичних умов у людей, що живуть в цих областях світу.
Відповідно, люди зі слабкими природними оборонами (без меланіну в їх шкірі) швидше за все, мають меланому. Наприклад, серед жителів Казахстану було восьме збільшення частоти меланома серед відвідувачів з білою шкірою порівняно з представниками корінного населення.
Статистика ВООЗ свідчить про те, що Чим вище природна кількість меланоцитів в шкірі, тим нижче ризик меланомиНевисока пігментація – це фактор ризику.
Оцінка цього фактора можливо вже з поточною клінічною експертизою. Люди, які належать до фенотипу, зазвичай слабо схильні до сонячних опіків і дуже схильні до розвитку сонячних опіків через високу чутливість до УФ-випромінювання, тому їх ризик розвитку меланоми подвійний.
Ще одним ознакою порушення рівня пігментації є наявність великої кількості веснянок на шкірі, які обумовлені накопиченням проліферуючих меланоцитів в базальному шарі епідермісу і епітелію зовнішніх частин волосяних фолікул.
Вкрай сильний ступінь пігментації, що характеризується повним відсутністю меланіну в організмі, спостерігається в альбіносі. Карциногенні ефекти УФ-випромінювання є настільки великим, що більшість людей розвиваються припустимими ураженнями або пухлинами шкіри у віці від 20 років, і так руйнівними, що менше 10% цих людей живуть до 30 років.
Знакові доріжки
На сьогоднішній день найбільш вивченими є наступні напрями сигналізації:
Відрізки починаються, коли вчені намагаються відслідковувати сувору специфіку шляху сигналізації і процес його регулює. Це помилка припустити, що активація цих шляхів сигналізації є ознакою неопластикової трансформації в меланомі.
По-перше, ці сигнальні доріжки існують, що регулюють диференціацію клітин в антенальних і післянатальних періодах, забезпечують відтворення впливу певних гормонів та інших типових процесів, спрямованих на збереження нормального функціонування клітин.
По-друге, ці шляхи беруть участь не тільки в меланоцитах: вони універсальні сигнальні системи, що беруть участь у більшості клітин організму людини. Що це таке? У будь-якому з сигнальних шляхів може виникнути одна або інша генетична або епігенетична поломка, результат якого буде негативним регулюванням сигналізації з результатом в карциногенезі.
МАПК
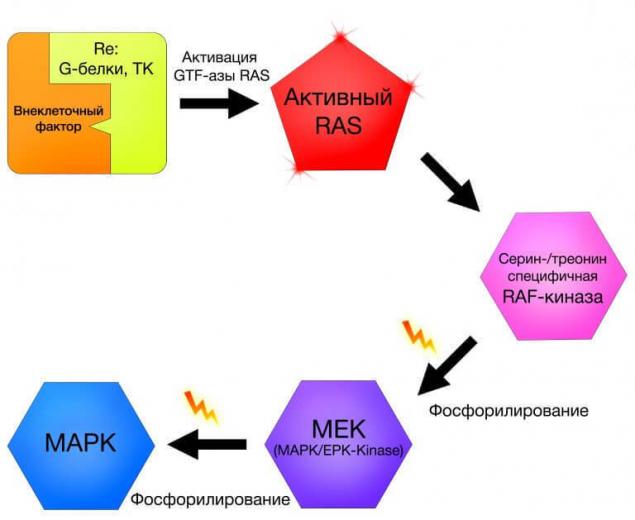
кластер MARK (мітоген-активований протеїнкінази) містить внутрішньоклітинні сигнальні доріжки широкого спектру функцій [8]. Критерієм включення в групу МАРК є наявність мітогенно-активованих протеїнкінази в модулі, які крім кінази, містять білкові фосфори та білкові колекціонери білкових допоміжних комплексів [9].
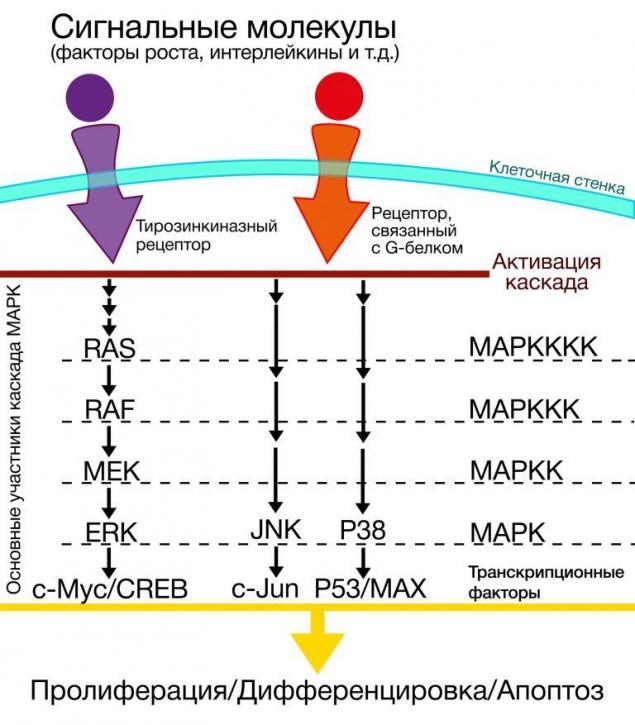
Незважаючи на те, що кластер МАРК не має особливого інтересу в контексті карциногенезу, але ті мутації, які відбуваються в генах білків, які беруть участь у сигналізації шляхів в клітинах меланоми шкіри, вимагають більш детального розгляду. Серед багатьох кінази, які беруть участь у передачі сигналу, є так звані кінази Raf в кластері MARK.
Raf- це сім'я сиринно-тероїнових кінази білків, ім'я якого є акронімом для швидкого прискорення Fibrosarcoma. Але де відбувається швидко зростаюча фібросаркома, і де знаходиться меланома шкіри?
Справа в тому, що в глибоких механізмах онкогенезу часто зустрічаються перехрестя, такі як в нашому випадку, в тому числі. У контексті даної статті немає сенсу розбухнути на детальну презентацію етапів онкогенезу фіброзою, тому ми зупинимося з особливостями расової активності в меланомі шкіри.
БРАТИ
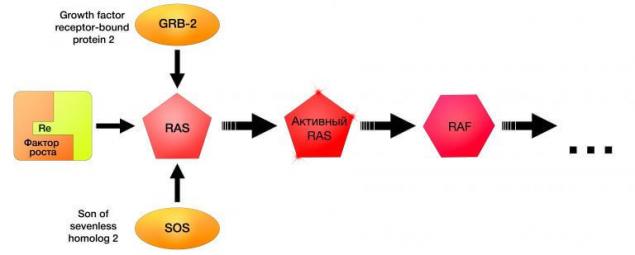
Відстеження в сім'ї Raf- для нас може бути B-raf, білок, який грає роль в каскаді MARK, наслідки яких є диференціація клітин і поділ. В-раф білок шифрується геном однойменної назви, яка буде абсолютно невизначеною, якщо не для одного «але»: що виникає під впливом надмірної ультрафіолетової опромінення в цьому гені V600[x] (на [x] в цьому випадку означає позначення не синонімної заміщення, що призводить до мутації), що складається з заміни люцину з лейцином (V600L), лізином (V600K) або глутаміновою кислотою (V600E) в 600-му положенні, служить стартером неопластичної трансформації в меланомі, а також цільовими препаратами VRA600. Це так званий "нібас": іміатин, сорб, вемурафеніб і т.д.
У рандомізованих клінічних дослідженнях було виявлено цікава особливість: якщо в перевіреному мутованій системі пацієнта BRAF, низькі молекулярні інгібітори BRAF привели до гальмування розвитку меланоми, то споживання «нібів» пацієнтами, мутації BRAF були сумнівні або не привели до мутацій іншої каскадної - RAS-RAF-MEK-ERK, патологічно активують її і ініціюваючи неопластикову трансформацію.
Таким чином, можна зробити висновок, що низькомолекулярні інгібітори BRAF, хоча вони є ефективним терапевтичним агентом, як і раніше показують небезпечну неоднозначність при недостатньому діагнозі або помилковому трактуванні стану BRAF пацієнта.
З 2011 року альтернатива хіміотерапії для пацієнтів з метастатичним меланом шкіри з мутаціями BRAF V600E - препарат vemurafenib (Zeloraf). У самому серці його дії є гальмування димеризації BRAF, оскільки у мутації V600E спостерігається збільшення сигналізації ERK в результаті занурення мутантної кінази.
У фазі я проваджу відповідь на vemurafenib було отримано у 81% хворих на меланому з мутацією V600E (по зменшенню діаметру всіх пухлинних вузлів на 30%). У фазі ІІІ проваджень загальний виживання протягом 6 місяців спостерігається в 84% пацієнтів з метастатичним меланомом, безперервне виживання було 5-7 місяців.
8135934
Однак у 20-30% пацієнтів, які отримують вемурафеніб, спостерігаються побічні ефекти: злоякісність доброякісних уражень і поява раку шкіри квуму, що асоціюється з активацією шляху МАПК через мутації РАС при при пригнічення РФ. Деякі пацієнти розробляють нові осередки меланоми з диким типом BRAF. Останні дослідження показують, що MARK може бути більш ефективно пригнічений при використанні не тільки BRAF (в монотерапевтичному режимі), але і інгібіторів MEK.
Ця комбінація є не тільки ефективнішою, вона запобігає розвитку стійкості до терапії, а також виникнення найбільш серйозного небажаного ефекту інгібіторів БРФ – онкологічних захворювань клітин.
Припустимо, що ефект може мати поєднання лікарських засобів сорбенту і лонафарнібу - інгібітора дальності (реакція, що веде до ініціації карциногенної активності білка RAS) і "розбір" RAS на клітинній мембрані.
У січні 2014 року для лікування меланоми було зареєстровано новий цільовий препарат dabrafenib. На відміну від vemurafenib, dabrafenib працює не тільки при заміні V600E, але і при мутації V600K. Інгібітори BRAF були першими препаратами, щоб мати ефект у пацієнтів з метастазами мозку. На жаль, майже всі пацієнти, які відповіли на вемурафеніб, розвиваються стійкістю до терапії.
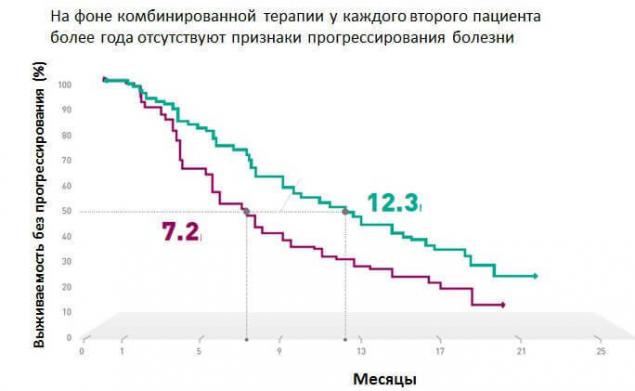
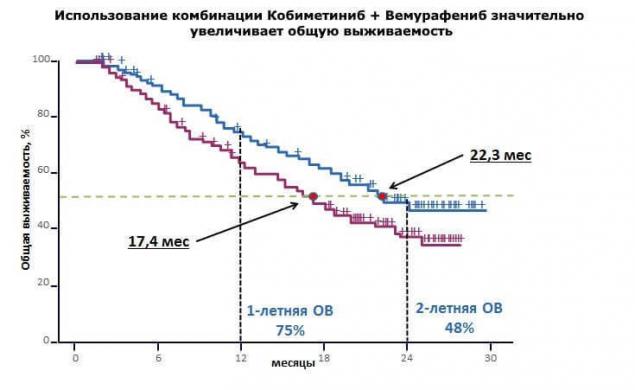
«Частина клінічного дослідження коБРІМ, підтверджена, що комбінована терапія з використанням vemurafenib і cobimetinib в 90% випадків досягає відповіді на терапія у пацієнтів з подвійною метастатичним меланом.» Один у двох пацієнтів не має ознак прогресування хвороби більше року і загальними виживаннями підходи до двох років. й
МЕК
MEK – це сиринно-трионова кінази кластера МАРК, активована завдяки змінам вищих посилань сигнальної мережі.
Роль MEK полягає в інтеграції сигналів з різних факторів росту з подальшою активацією проліферації. В рамках каскаду МАРК МЕК бере участь у регулюванні діяльності транскрипційних чинників, таких як С-мик.
Протеїн Мик зашифрований геном тієї ж назви і є прото-онкогеном не тільки «канонічним» прикладом фактора транскриптора, але і контролює структуру хроматину шляхом регулювання ацетиляції гітона, що в свою чергу впливає на активність експресії гена. мутантний ген м'язів, розташований на хромосомі 8, зустрічається в багатьох типах пухлин; класичний приклад - транслокація т (8;14), що веде до виникнення і розвитку лімфоми Буркету.
Експерименти, спрямовані на пригнічення його привели до аналізу пухлинних клітин легенів у мишей. Отже, музи є лікувальною метою, зокрема, розвитком селекційних алеостеричних інгібіторів м'язів є актуальним для клінічних досліджень.
НРАС
N-RAS (Neuroblastoma-Ras) є кодування гена білків однойменної назви, яка входить до складу так званої суперфіміської рози – невеликого GTP-az. Разом з NRAS ця родина включає гени K-Ras, H-Ras (що викликає неопластичну трансформацію, коли Kristen і Harvey контракт сармавірус, відповідно) та інших. На сьогоднішній день НРАС був визначений як передача проліферативних сигналів від рецепторів фактора росту. НРАС, як і вся суперсімія, бере участь у ходовому шляху сигналізації МАРК.
У 10-26% випадків меланоми шкіри, переважно опромінюють УФ. Пригнічення НРАС відкриває нові горизонти в меланоматерапії, демонструючи на практиці важливість тісного зв’язку всіх зв’язків в умовах сигналізації. Перший препарат для цільової терапії меланоми з мутацією NRAS - інгібітор MEK162, завдяки якому майже 70% пацієнтів в даній категорії вдається отримати відповідь на лікування і домогтися стабілізації захворювання.
c-КІТ
Останнє посилання в каскаді МАРК розглядається в контексті цієї статті буде ген c-KIT. Останній, але не менше, цей ген, який є прото-oncogene, протеїн якого є рецептором факторів росту стовбурових клітин, виявляється в кожному третій випадок меланоми різних локалізацій: як шкіри, так і слизових меланоми.
709550 р.
Схема сигналу Kit
Що стосується багатьох інших мутацій, існують препарати, спрямовані на гальмування мутацій Kit. З огляду на неперервну домішку зв’язків шляхової доріжки MARK, не дивно, що мутація Kit успішно пригнічується вже відомою цільовою терапією BRAF-позитивних меланоми "-нібас": Іманіб, нілатиніб та інші препарати.
PI3K – Фосфінозитол-3-Кинази шлях
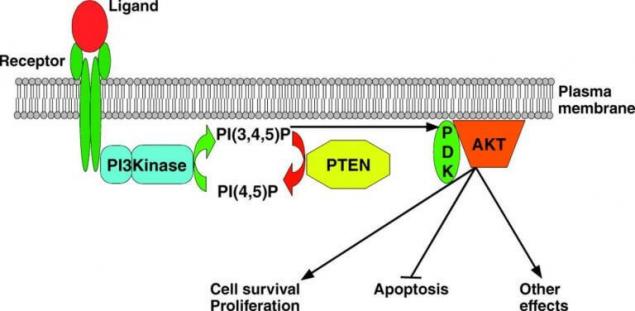
Наступним компонентом проліферативного сигналу патологічно активовано в клітинах меланоми є PI3K, фосфатиноситол-3-кінази, часто згадується в літературі як PI3K/AKT/mTOR. Це каскад реакцій, основні події яких розгортаються навколо наступних ферментів: фосфатиносиди-3-кінази (ПІ3К), сімейство білкових кінази B, компоненти яких є сиринно-тероїнової кінази АКТ 1, 2, 3 та іншої сиринно-трионової кінази, що називається «таргетом раamицину» і позначений як МТОР.
Цей шлях сигналізації є універсальним для більшості клітин людини і забезпечує розмноження клітин, обмін речовин і диференціацію.
В умовах карциногенезу в клітинах меланоми шкіри, компоненти АКТ і ПЕН є найбільшим інтересом.
 Р
Р
Класичний Шлях PTEN
ПЕН
PTEN - це ферментна фосфатаза, яка є негативним регулятором шляхової дороги PI3K і шифрується геном онкосупресора тієї ж назви. За подразненням фосфатної групи в фосфатидилінозитол-3-фосфати, цей фермент гальмує (до повного блокаду) провідність сигналу вздовж фосфатинозитол-3-кінази шляху. Інактивація ТЕН міститься в багатьох пухлинах, оскільки це призводить до неконтрольованого поділу з втратою диференціації, порушення метаболізму клітин і перевернутого синтезу. У 10-30% меланоми знайдено інактивуючі мутації PTEN.
АКТИВНІСТЬ
Ще один компонент сигналізації PI3K, розглянутий для розвитку меланоми, буде сімейство АКТ.
Сім'я АКТ є групою кінази, які прикріплюють залишки фосфорної кислоти до різних білків цитозолу, контроль їх активності. Ця сім'я виконує як класичні функції, наприклад, регулювання проліферації, диференціації та зміни цитоскелетону, так і делікатно в контексті функцій карциногенезу ангіогенезу, уникнення апоптозу та придбання опору до цитостатиків.
Сім'я ACT включає в себе 3 підвиди протеїнкінази: ACT-1, ACT-2 і ACT-3, які є продуктами відповідних генів.
Акт1 - α-серин / кінази білків триеоніну - пригнічення апоптозу, біосинтезу білка (в напрямку «плюсної тканини», гіпертрофії міоцитів і т.д.).
Акт2 - ß -серин / кінази білків триеоніну - бере участь в обміні інсуліну, індукує транспортування глюкози, що здійснюється GLUT-4.
Акт3 - γ-серин/трионін-протеїнкіназа - функція надійно невідома.
Різні види АКТ перекопчені в 45-70% меланоми. Експерименти, що проводяться з фосфатазом PHLPP, які є каталізатором для дефосориляції в молекулі АКТ-1, а також інактивації АКТ-1, привели до апоптозу та уповільненого розмноження в неопластично змінених клітинах.
МАР
Цей протеїнкінази також показує специфічність триеонін-серину, що існує у вигляді двох внутрішньоклітинних комплексів: TORC1 і TORC2. Мета рапаміцину – перший комплекс, який є лікувальною метою імуносупресора тієї ж назви.
Але ссавціанська мета Rapamycin - це не мета rapamycin, але компонент PI3K сигнального шляху, який грає ключову роль в клітинному росту. Незважаючи на те, що молекула TOR є невидимою похідною його двох підпунктів, останні мають дуже різні потужності.
Пошкодження першого комплексу не має ніяких катастрофічних наслідків для клітинки, а порушення TOR2 призведе до так званого «підлокітника» клітинного циклу в фазі G2 / M після декількох поколінь. При ураженні обох підпунктів клітинка замерзає в фазі G0 в наступному генеруванні. Таким чином, він стає зрозумілим, чи самостійно або разом з TOR1, але якось TOR2 має сильний вплив на цикл клітин.
У статті розглянуто наступні компоненти системи керування циклом клітинок, розглянуті CDKN2A, CDK4 і CDK6 системи цикллінзасе. Мутации, вилучення, гіперметика промотора - це розлади, виявлені в 50% (CDKN2A) і 10-12% (CDK4) випадків меланоми, а спадкові гермінальні мутації в генах схильності може викликати фміліальну меланому в 15% випадків.
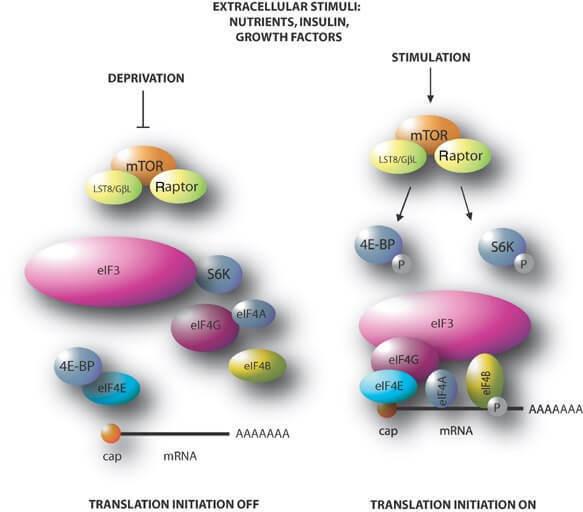
Рисунок 1 Механізм дії рапаміцину
CDKN2A (cyclin-залежні kinase Inhibitor 2A) є геном, який локалізований в короткій руці хромосоми 9 і кодує два білки, які є онкосупресорами: p14 і p16. За допомогою цих білків активність, мабуть, найвідомішого онкосупресора p53 і ретинобластома білка регулюється.
У разі мутації CDKN2A (застосування кодону R112), уражені обидва продукти гена. Цікаво, що ця мутація є домінуючою у випадках спадкової меланоми в сім'ях скандинавських національностей. У 30-40% випадків меланоми, особливо в контексті фміліальної меланоми. Ген-поліморфізм важливий при визначенні частоти розвитку пухлин і його стійкості до поліхіміотерапії.
Тим не менш, це неправильно припустити, що CDKN2A є виключно відповідальним за фамілі випадки. Споричні соматичні мутації CDKN2A виникають в 50% випадків меланоми шкіри, реалізовані або шляхом деактивації p16 або шляхом метилювання промотора (10%).
Білковий комплекс cyclin-D та CDK4 регулює експресію протеїну ретинобластома, що впливає на активність клітинного циклу та проліферацію клітин. Каскад реакцій нижче схематично показує послідовність подій в генетичному апараті і сам клітинку, коли вищі нормативні механізми, в цьому випадку білок p16, зламається:
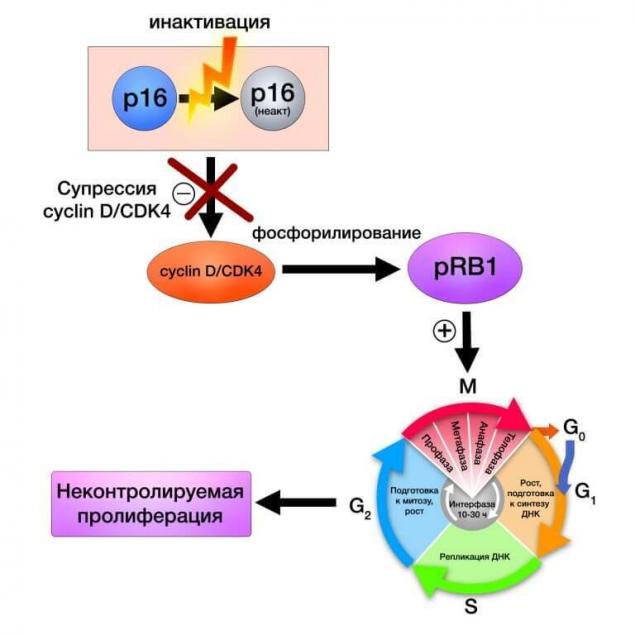
CDK4 (cyclin-залежні кінази 4) є ферментом, зашифрованим геном однойменної назви і є частиною сімейства циліндро-залежних кінази. В рамках єдиної системи керування циклом клітин CDK4, як CDK6, обмежується відповідними білками та білками ретинобластома.
У контексті карциногенезу ці елементи діють як важелі для контролінгу клітинного циклу, неконтрольована активність яких може бути результатом певних порушень в ферментах управління. CDK як молекулярна ціль є інтересом не тільки до дерматокологів, так як інгібітори CDK в цілому клас лікарських засобів протиракових препаратів широко використовуються для лікування широкого спектру типів раку, таких як естроген-позитивний і HER-негативний рак груди, лейкемія, рак щитоподібної залози (палоциклліб, глибин, абемацліб).
Циклінозалежні інгібітори кінази блокують активність відповідного ферменту як самостійно, так і в ферменті + цикллінний комплекс; ці препарати діють, як правило, в фазі клітинного циклу G1. Зменшення активності CDK може бути досягнуто, наприклад, зниження рівня експресії генів циклінів або збільшення ступеня розбиття циклінів.
інгібітори CDK не тільки забезпечують достатнє зростання клітин, але і мають можливість зупинити цикл на стадії G1 у відповідь на пошкодження ДНК, несприятливі зовнішні умови тощо.
ВНТ
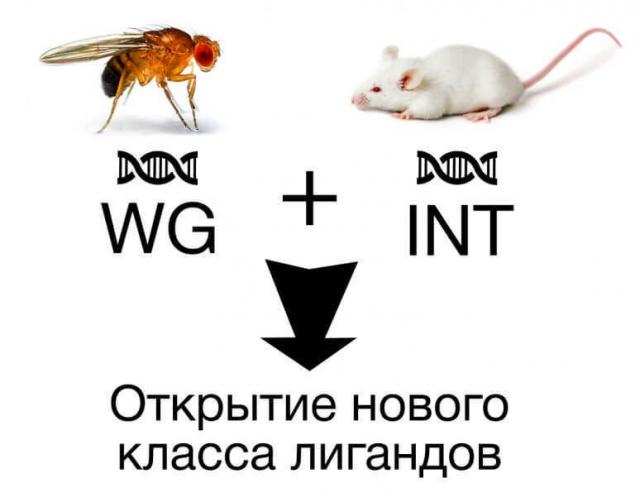
Каскад WNT - це приклад класичного напряму сигналізації, що регулює проліферативну активність та диференціацію клітин. Цей шлях отримав свою назву від злиття двох генів: WG Drosophila летить, які пригнічували зростання крила, і INT, які спочатку розглядалися в дослідження раку грудей в мишей. В одній структурі білок поєднаний на основі ряду функцій, що дозволило відрізняти новий клас лігандів.
У своїй структурі WNT є комплексний комплекс, з графічним зображенням якого можна помітити схожість з відкритою долонею. Слід зазначити, що рівень глікозиляції не впливає на секреторну активність даного комплексу, але Н-глікозин збільшує секрецію ВНТ.
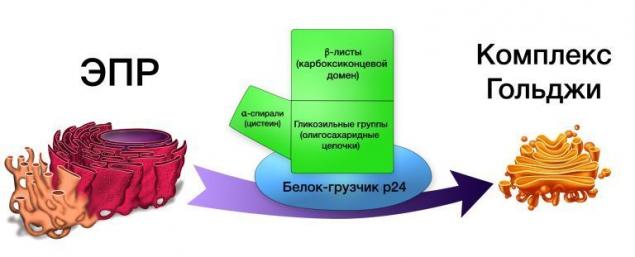
Білки WNT виводяться білками комплексних Golgi, таких як GPR177, і переносять на поверхню клітин, використовуючи білки p24 навантажувача.
WNT може діяти на клітинку відповідно до канонічного або неканонічного шляху, вони також β-катенін залежний і β-катенін незалежного шляху, відповідно. У першому випадку концентрація клітинних β-катенінових змін, кінцеве посилання якого є контрольом генетичного виразу, що впливає на морфогенез клітин, а в другому випадку полярність клітини регулюється шляхом стимулювання реорганізації цитоскелетона і зміни інтенсивності метаболізму кальцію.
Канонічний шлях – це послідовність реакцій, спрямованих на накопичення ß-catenin в клітині та її безпосередню доставку до нуклесу, де він буде діяти як транскрипційний фактор.
Шлях WNT починається з підключення WNT до рецептора для ліпопротеїну низької щільності (Low-density lipoprotein Relater протеїн-5, LRP-5), потім шляхом активування ефекту білка AXIN1 APC (негативний регулятор концентрації ß-catenin, мутації гена, що призводять до виникнення і розвитку кольорового раку), кінази GSK3, CK1 (кінази глікогедж).
Джерело: medach.pro/surgery/onkologiya-hirurgicheskie-distiplinyi/melanoma/
Маланома шкіри є результатом неопластичної трансформації меланоцитів – клітин, які виробляють різні варіації пігментного меланіну. Меланоцити виростають з нервового гребеня - група клітин маргінальної частини нервової кори.

Нервові клітини мають високий рівень міграції, а більшість з них спрямовуються з нервової труби, що утворюють різні нервові та ендокринні структури. Тим не менш, меланобласти показують свою унікальність навіть перед потенційним придбанням пухлинного фенотипу і мігрувати не внизу вгору, але далеко від нервової труби.
Молекулярні механізми цього процесу досі невідомі. Доля меланоблапласту навряд чи може бути викликана заздалегідь: цей елемент може згодом розвиватися в нейрон, лейоміоцит, а в разі високого рівня меланоцито-гліального потенціалу - і в компоненту глі.
Після диференціації в меланоциті, тіло (сома) і процеси виділяються, які розташовані в базальних і шипкових шарах епідермісу, відповідно. Під дією меланоцимуляційних і адренакортикотропних гормонів, а також сонячних променів, меаносом синтезу (ев)меланін (і пекомланін), функція якого полягає в тому, щоб захистити ядерний апарат клітин від пошкодження ультрафіолетового випромінювання.
Сингабаритний меланінов перевозиться в спінний шар епідермісу уздовж процесів меланоциту, потім в кератиноцити епідермісу, що дає шкірі тан. Після деякого часу цей полімер гідролізується в лізосом, і шкіра повертає свій звичний відтінок.
Цей сценарій можна назвати ідеальним. Але що відбувається для тих, хто менш завадити? Ключ до ініціювання неопластичної трансформації меланоциту – це нелетична мутація в її генетичному апараті. Причиною такого пошкодження може бути різні ефекти: як фізичні, так і хімічні.
Як при більшості новоутворень причини онкогенезу в меланомі можна поєднувати за кількома ознаками:
1) Пригнічення активності генів онкосупресора;
2) Активація прото-oncogenes;
(3) Екзогенні мутації, отримані в різних напрямках.
Що таке меланома? Здавалося б, що зовнішня локалізація, відомі алгоритми на стадії самовизначення, висока імуногенність пухлини повинна максимально ефективно проводити її терапія.
Однак непередбачуваність метастатичного каскаду, пов'язаної з відсутністю спеціально розробленої терапії метастатичної меланоми, непередбачуваності клінічних і морфологічних форм - як мікро-, так і макроскопічної - і найвищою частотою рецидиву призводять до того, що сьогодні захворюваність меланоми зростає в усіх країнах світу; рівень медіанності меланоми не більше 8 місяців, а ефективність радикального хірургічного лікування не перевищує 65%. Відповіді можна відповісти тільки клініки та пацієнти.
Можливість занурення геному дозволили нам проникнути в розуміння карциногенезу в меланомі шкіри. Дослідження процесів, які ініціюють трансформацію меланоцитів в найбільш агресивні існуючі пухлини, є найважливішим кроком у дослідженні не тільки меланоми шкіри, але й інших пухлин.
Тільки шляхом виявлення нових цілей можна призначати цільову терапію, розширити діагностичні горизонти, а також переглянути класифікацію за допомогою клініко-морфологічних форм з перевагами поєднання підвидів меланоми на основі природи мутацій. З цим на увазі слід звертати особливу увагу огляд механізмів карциногенезу в меланомі.
Одним з найважливіших досягнень у дослідженні карциногенезу в меланомі є аналіз механізмів передачі сигналів, які активуються під час його розвитку. На основі етіологічного фактора виявлення певних мутацій в меланомі шкіри може відповісти питання взаємозв’язків етіології та патогенезу, клінічних форм і терапевтичних рішень.
Що таке сигнал? Це свого роду «релей», в якому в строго визначеному порядку бере участь послідовність певних молекул, які беруть участь у передачі будь-якого виду сигналу від рецептора клітин в клітинку. Шлях сигналізації може бути активований екзогенно—розривні чинники, нейротрансмітери, гормони — або ендогенно—за сигналами стартера, активовані з різних причин в клітині.
епідеміологія
Середній вік пацієнтів меланоми становить близько 45 років, але в останні роки меланома стала все більш поширеною в дуже молодих людей (15-25 років). Ця пухлина зустрічається у чоловіків і жінок, і у жінок 1,5-2 рази частіше. За статистикою, для кожного 100 000 здорових людей, є 14 пацієнтів з меланоми.
У 2006 році в Росії зареєстровано 7364 нових хворих на меланоми шкіри та 3,033 смерті. Річна кількість випадків меланоми в Росії становить 5,700 осіб, 2,200 з яких, ймовірно, відмирають від захворювання.
У різних регіонах світу значно змінюється захворюваність меланоми. Згідно з ВООЗ, між 1988 та 2012 року, найвищий стандартизований захворюваність меланоми шкіри (25–29.8o/ooh) серед білих популяцій Австралії та Нової Зеландії. Серед європейців, які живуть в Зімбабве, білі чоловіки США (Лес-Анджелес, Сан-Франциско), жінки Австрії, Норвегії.
Високий рівень 8,8-14.1 o /ooh був серед жителів Данії, Італії, Швейцарії, Швеції, чоловіків Австрії та Норвегії. Найнижчий стандартизований захворюванні меланоми шкіри 0,1-1,5 ° /ooh виявлений в Алжирі, в індійських і чорних жителів США, Уганда, Зімбабве, Китай, Корея, Японія.
Поява меланоми шкіри також впливає на расу людини. Згідно з статистикою ВООЗ, чорношкірі в 4 рази рідше страждають від меланоми, ніж кавказький. Існує припущення, що темніше шкіра містить більше меланіну в епідермісі, що сприяє більшій затримці в УФ-випромінюванні, і таким чином є більш надійним захистом від шкідливого ефекту на меланоцитах, розробленої еволюціонально відповідно до кліматичних умов у людей, що живуть в цих областях світу.
Відповідно, люди зі слабкими природними оборонами (без меланіну в їх шкірі) швидше за все, мають меланому. Наприклад, серед жителів Казахстану було восьме збільшення частоти меланома серед відвідувачів з білою шкірою порівняно з представниками корінного населення.
Статистика ВООЗ свідчить про те, що Чим вище природна кількість меланоцитів в шкірі, тим нижче ризик меланомиНевисока пігментація – це фактор ризику.
Оцінка цього фактора можливо вже з поточною клінічною експертизою. Люди, які належать до фенотипу, зазвичай слабо схильні до сонячних опіків і дуже схильні до розвитку сонячних опіків через високу чутливість до УФ-випромінювання, тому їх ризик розвитку меланоми подвійний.
Ще одним ознакою порушення рівня пігментації є наявність великої кількості веснянок на шкірі, які обумовлені накопиченням проліферуючих меланоцитів в базальному шарі епідермісу і епітелію зовнішніх частин волосяних фолікул.
Вкрай сильний ступінь пігментації, що характеризується повним відсутністю меланіну в організмі, спостерігається в альбіносі. Карциногенні ефекти УФ-випромінювання є настільки великим, що більшість людей розвиваються припустимими ураженнями або пухлинами шкіри у віці від 20 років, і так руйнівними, що менше 10% цих людей живуть до 30 років.
Знакові доріжки
На сьогоднішній день найбільш вивченими є наступні напрями сигналізації:
- Марк.
- ПІ3К
- Cайт
- Не
Відрізки починаються, коли вчені намагаються відслідковувати сувору специфіку шляху сигналізації і процес його регулює. Це помилка припустити, що активація цих шляхів сигналізації є ознакою неопластикової трансформації в меланомі.
По-перше, ці сигнальні доріжки існують, що регулюють диференціацію клітин в антенальних і післянатальних періодах, забезпечують відтворення впливу певних гормонів та інших типових процесів, спрямованих на збереження нормального функціонування клітин.
По-друге, ці шляхи беруть участь не тільки в меланоцитах: вони універсальні сигнальні системи, що беруть участь у більшості клітин організму людини. Що це таке? У будь-якому з сигнальних шляхів може виникнути одна або інша генетична або епігенетична поломка, результат якого буде негативним регулюванням сигналізації з результатом в карциногенезі.
МАПК
кластер MARK (мітоген-активований протеїнкінази) містить внутрішньоклітинні сигнальні доріжки широкого спектру функцій [8]. Критерієм включення в групу МАРК є наявність мітогенно-активованих протеїнкінази в модулі, які крім кінази, містять білкові фосфори та білкові колекціонери білкових допоміжних комплексів [9].

Незважаючи на те, що кластер МАРК не має особливого інтересу в контексті карциногенезу, але ті мутації, які відбуваються в генах білків, які беруть участь у сигналізації шляхів в клітинах меланоми шкіри, вимагають більш детального розгляду. Серед багатьох кінази, які беруть участь у передачі сигналу, є так звані кінази Raf в кластері MARK.
Raf- це сім'я сиринно-тероїнових кінази білків, ім'я якого є акронімом для швидкого прискорення Fibrosarcoma. Але де відбувається швидко зростаюча фібросаркома, і де знаходиться меланома шкіри?
Справа в тому, що в глибоких механізмах онкогенезу часто зустрічаються перехрестя, такі як в нашому випадку, в тому числі. У контексті даної статті немає сенсу розбухнути на детальну презентацію етапів онкогенезу фіброзою, тому ми зупинимося з особливостями расової активності в меланомі шкіри.
БРАТИ
Відстеження в сім'ї Raf- для нас може бути B-raf, білок, який грає роль в каскаді MARK, наслідки яких є диференціація клітин і поділ. В-раф білок шифрується геном однойменної назви, яка буде абсолютно невизначеною, якщо не для одного «але»: що виникає під впливом надмірної ультрафіолетової опромінення в цьому гені V600[x] (на [x] в цьому випадку означає позначення не синонімної заміщення, що призводить до мутації), що складається з заміни люцину з лейцином (V600L), лізином (V600K) або глутаміновою кислотою (V600E) в 600-му положенні, служить стартером неопластичної трансформації в меланомі, а також цільовими препаратами VRA600. Це так званий "нібас": іміатин, сорб, вемурафеніб і т.д.
У рандомізованих клінічних дослідженнях було виявлено цікава особливість: якщо в перевіреному мутованій системі пацієнта BRAF, низькі молекулярні інгібітори BRAF привели до гальмування розвитку меланоми, то споживання «нібів» пацієнтами, мутації BRAF були сумнівні або не привели до мутацій іншої каскадної - RAS-RAF-MEK-ERK, патологічно активують її і ініціюваючи неопластикову трансформацію.
Таким чином, можна зробити висновок, що низькомолекулярні інгібітори BRAF, хоча вони є ефективним терапевтичним агентом, як і раніше показують небезпечну неоднозначність при недостатньому діагнозі або помилковому трактуванні стану BRAF пацієнта.
З 2011 року альтернатива хіміотерапії для пацієнтів з метастатичним меланом шкіри з мутаціями BRAF V600E - препарат vemurafenib (Zeloraf). У самому серці його дії є гальмування димеризації BRAF, оскільки у мутації V600E спостерігається збільшення сигналізації ERK в результаті занурення мутантної кінази.
У фазі я проваджу відповідь на vemurafenib було отримано у 81% хворих на меланому з мутацією V600E (по зменшенню діаметру всіх пухлинних вузлів на 30%). У фазі ІІІ проваджень загальний виживання протягом 6 місяців спостерігається в 84% пацієнтів з метастатичним меланомом, безперервне виживання було 5-7 місяців.
8135934
Однак у 20-30% пацієнтів, які отримують вемурафеніб, спостерігаються побічні ефекти: злоякісність доброякісних уражень і поява раку шкіри квуму, що асоціюється з активацією шляху МАПК через мутації РАС при при пригнічення РФ. Деякі пацієнти розробляють нові осередки меланоми з диким типом BRAF. Останні дослідження показують, що MARK може бути більш ефективно пригнічений при використанні не тільки BRAF (в монотерапевтичному режимі), але і інгібіторів MEK.
Ця комбінація є не тільки ефективнішою, вона запобігає розвитку стійкості до терапії, а також виникнення найбільш серйозного небажаного ефекту інгібіторів БРФ – онкологічних захворювань клітин.
Припустимо, що ефект може мати поєднання лікарських засобів сорбенту і лонафарнібу - інгібітора дальності (реакція, що веде до ініціації карциногенної активності білка RAS) і "розбір" RAS на клітинній мембрані.
У січні 2014 року для лікування меланоми було зареєстровано новий цільовий препарат dabrafenib. На відміну від vemurafenib, dabrafenib працює не тільки при заміні V600E, але і при мутації V600K. Інгібітори BRAF були першими препаратами, щоб мати ефект у пацієнтів з метастазами мозку. На жаль, майже всі пацієнти, які відповіли на вемурафеніб, розвиваються стійкістю до терапії.

«Частина клінічного дослідження коБРІМ, підтверджена, що комбінована терапія з використанням vemurafenib і cobimetinib в 90% випадків досягає відповіді на терапія у пацієнтів з подвійною метастатичним меланом.» Один у двох пацієнтів не має ознак прогресування хвороби більше року і загальними виживаннями підходи до двох років. й

МЕК
MEK – це сиринно-трионова кінази кластера МАРК, активована завдяки змінам вищих посилань сигнальної мережі.
Роль MEK полягає в інтеграції сигналів з різних факторів росту з подальшою активацією проліферації. В рамках каскаду МАРК МЕК бере участь у регулюванні діяльності транскрипційних чинників, таких як С-мик.

Протеїн Мик зашифрований геном тієї ж назви і є прото-онкогеном не тільки «канонічним» прикладом фактора транскриптора, але і контролює структуру хроматину шляхом регулювання ацетиляції гітона, що в свою чергу впливає на активність експресії гена. мутантний ген м'язів, розташований на хромосомі 8, зустрічається в багатьох типах пухлин; класичний приклад - транслокація т (8;14), що веде до виникнення і розвитку лімфоми Буркету.
Експерименти, спрямовані на пригнічення його привели до аналізу пухлинних клітин легенів у мишей. Отже, музи є лікувальною метою, зокрема, розвитком селекційних алеостеричних інгібіторів м'язів є актуальним для клінічних досліджень.
НРАС
N-RAS (Neuroblastoma-Ras) є кодування гена білків однойменної назви, яка входить до складу так званої суперфіміської рози – невеликого GTP-az. Разом з NRAS ця родина включає гени K-Ras, H-Ras (що викликає неопластичну трансформацію, коли Kristen і Harvey контракт сармавірус, відповідно) та інших. На сьогоднішній день НРАС був визначений як передача проліферативних сигналів від рецепторів фактора росту. НРАС, як і вся суперсімія, бере участь у ходовому шляху сигналізації МАРК.

У 10-26% випадків меланоми шкіри, переважно опромінюють УФ. Пригнічення НРАС відкриває нові горизонти в меланоматерапії, демонструючи на практиці важливість тісного зв’язку всіх зв’язків в умовах сигналізації. Перший препарат для цільової терапії меланоми з мутацією NRAS - інгібітор MEK162, завдяки якому майже 70% пацієнтів в даній категорії вдається отримати відповідь на лікування і домогтися стабілізації захворювання.
c-КІТ
Останнє посилання в каскаді МАРК розглядається в контексті цієї статті буде ген c-KIT. Останній, але не менше, цей ген, який є прото-oncogene, протеїн якого є рецептором факторів росту стовбурових клітин, виявляється в кожному третій випадок меланоми різних локалізацій: як шкіри, так і слизових меланоми.
709550 р.
Схема сигналу Kit
Що стосується багатьох інших мутацій, існують препарати, спрямовані на гальмування мутацій Kit. З огляду на неперервну домішку зв’язків шляхової доріжки MARK, не дивно, що мутація Kit успішно пригнічується вже відомою цільовою терапією BRAF-позитивних меланоми "-нібас": Іманіб, нілатиніб та інші препарати.
PI3K – Фосфінозитол-3-Кинази шлях
Наступним компонентом проліферативного сигналу патологічно активовано в клітинах меланоми є PI3K, фосфатиноситол-3-кінази, часто згадується в літературі як PI3K/AKT/mTOR. Це каскад реакцій, основні події яких розгортаються навколо наступних ферментів: фосфатиносиди-3-кінази (ПІ3К), сімейство білкових кінази B, компоненти яких є сиринно-тероїнової кінази АКТ 1, 2, 3 та іншої сиринно-трионової кінази, що називається «таргетом раamицину» і позначений як МТОР.
Цей шлях сигналізації є універсальним для більшості клітин людини і забезпечує розмноження клітин, обмін речовин і диференціацію.
В умовах карциногенезу в клітинах меланоми шкіри, компоненти АКТ і ПЕН є найбільшим інтересом.
 Р
РКласичний Шлях PTEN
ПЕН
PTEN - це ферментна фосфатаза, яка є негативним регулятором шляхової дороги PI3K і шифрується геном онкосупресора тієї ж назви. За подразненням фосфатної групи в фосфатидилінозитол-3-фосфати, цей фермент гальмує (до повного блокаду) провідність сигналу вздовж фосфатинозитол-3-кінази шляху. Інактивація ТЕН міститься в багатьох пухлинах, оскільки це призводить до неконтрольованого поділу з втратою диференціації, порушення метаболізму клітин і перевернутого синтезу. У 10-30% меланоми знайдено інактивуючі мутації PTEN.
АКТИВНІСТЬ
Ще один компонент сигналізації PI3K, розглянутий для розвитку меланоми, буде сімейство АКТ.
Сім'я АКТ є групою кінази, які прикріплюють залишки фосфорної кислоти до різних білків цитозолу, контроль їх активності. Ця сім'я виконує як класичні функції, наприклад, регулювання проліферації, диференціації та зміни цитоскелетону, так і делікатно в контексті функцій карциногенезу ангіогенезу, уникнення апоптозу та придбання опору до цитостатиків.
Сім'я ACT включає в себе 3 підвиди протеїнкінази: ACT-1, ACT-2 і ACT-3, які є продуктами відповідних генів.
Акт1 - α-серин / кінази білків триеоніну - пригнічення апоптозу, біосинтезу білка (в напрямку «плюсної тканини», гіпертрофії міоцитів і т.д.).
Акт2 - ß -серин / кінази білків триеоніну - бере участь в обміні інсуліну, індукує транспортування глюкози, що здійснюється GLUT-4.
Акт3 - γ-серин/трионін-протеїнкіназа - функція надійно невідома.
Різні види АКТ перекопчені в 45-70% меланоми. Експерименти, що проводяться з фосфатазом PHLPP, які є каталізатором для дефосориляції в молекулі АКТ-1, а також інактивації АКТ-1, привели до апоптозу та уповільненого розмноження в неопластично змінених клітинах.
МАР
Цей протеїнкінази також показує специфічність триеонін-серину, що існує у вигляді двох внутрішньоклітинних комплексів: TORC1 і TORC2. Мета рапаміцину – перший комплекс, який є лікувальною метою імуносупресора тієї ж назви.
Але ссавціанська мета Rapamycin - це не мета rapamycin, але компонент PI3K сигнального шляху, який грає ключову роль в клітинному росту. Незважаючи на те, що молекула TOR є невидимою похідною його двох підпунктів, останні мають дуже різні потужності.
Пошкодження першого комплексу не має ніяких катастрофічних наслідків для клітинки, а порушення TOR2 призведе до так званого «підлокітника» клітинного циклу в фазі G2 / M після декількох поколінь. При ураженні обох підпунктів клітинка замерзає в фазі G0 в наступному генеруванні. Таким чином, він стає зрозумілим, чи самостійно або разом з TOR1, але якось TOR2 має сильний вплив на цикл клітин.
У статті розглянуто наступні компоненти системи керування циклом клітинок, розглянуті CDKN2A, CDK4 і CDK6 системи цикллінзасе. Мутации, вилучення, гіперметика промотора - це розлади, виявлені в 50% (CDKN2A) і 10-12% (CDK4) випадків меланоми, а спадкові гермінальні мутації в генах схильності може викликати фміліальну меланому в 15% випадків.

Рисунок 1 Механізм дії рапаміцину
CDKN2A (cyclin-залежні kinase Inhibitor 2A) є геном, який локалізований в короткій руці хромосоми 9 і кодує два білки, які є онкосупресорами: p14 і p16. За допомогою цих білків активність, мабуть, найвідомішого онкосупресора p53 і ретинобластома білка регулюється.
У разі мутації CDKN2A (застосування кодону R112), уражені обидва продукти гена. Цікаво, що ця мутація є домінуючою у випадках спадкової меланоми в сім'ях скандинавських національностей. У 30-40% випадків меланоми, особливо в контексті фміліальної меланоми. Ген-поліморфізм важливий при визначенні частоти розвитку пухлин і його стійкості до поліхіміотерапії.
Тим не менш, це неправильно припустити, що CDKN2A є виключно відповідальним за фамілі випадки. Споричні соматичні мутації CDKN2A виникають в 50% випадків меланоми шкіри, реалізовані або шляхом деактивації p16 або шляхом метилювання промотора (10%).
Білковий комплекс cyclin-D та CDK4 регулює експресію протеїну ретинобластома, що впливає на активність клітинного циклу та проліферацію клітин. Каскад реакцій нижче схематично показує послідовність подій в генетичному апараті і сам клітинку, коли вищі нормативні механізми, в цьому випадку білок p16, зламається:

CDK4 (cyclin-залежні кінази 4) є ферментом, зашифрованим геном однойменної назви і є частиною сімейства циліндро-залежних кінази. В рамках єдиної системи керування циклом клітин CDK4, як CDK6, обмежується відповідними білками та білками ретинобластома.
У контексті карциногенезу ці елементи діють як важелі для контролінгу клітинного циклу, неконтрольована активність яких може бути результатом певних порушень в ферментах управління. CDK як молекулярна ціль є інтересом не тільки до дерматокологів, так як інгібітори CDK в цілому клас лікарських засобів протиракових препаратів широко використовуються для лікування широкого спектру типів раку, таких як естроген-позитивний і HER-негативний рак груди, лейкемія, рак щитоподібної залози (палоциклліб, глибин, абемацліб).
Циклінозалежні інгібітори кінази блокують активність відповідного ферменту як самостійно, так і в ферменті + цикллінний комплекс; ці препарати діють, як правило, в фазі клітинного циклу G1. Зменшення активності CDK може бути досягнуто, наприклад, зниження рівня експресії генів циклінів або збільшення ступеня розбиття циклінів.
інгібітори CDK не тільки забезпечують достатнє зростання клітин, але і мають можливість зупинити цикл на стадії G1 у відповідь на пошкодження ДНК, несприятливі зовнішні умови тощо.
ВНТ
Каскад WNT - це приклад класичного напряму сигналізації, що регулює проліферативну активність та диференціацію клітин. Цей шлях отримав свою назву від злиття двох генів: WG Drosophila летить, які пригнічували зростання крила, і INT, які спочатку розглядалися в дослідження раку грудей в мишей. В одній структурі білок поєднаний на основі ряду функцій, що дозволило відрізняти новий клас лігандів.

У своїй структурі WNT є комплексний комплекс, з графічним зображенням якого можна помітити схожість з відкритою долонею. Слід зазначити, що рівень глікозиляції не впливає на секреторну активність даного комплексу, але Н-глікозин збільшує секрецію ВНТ.

Білки WNT виводяться білками комплексних Golgi, таких як GPR177, і переносять на поверхню клітин, використовуючи білки p24 навантажувача.
WNT може діяти на клітинку відповідно до канонічного або неканонічного шляху, вони також β-катенін залежний і β-катенін незалежного шляху, відповідно. У першому випадку концентрація клітинних β-катенінових змін, кінцеве посилання якого є контрольом генетичного виразу, що впливає на морфогенез клітин, а в другому випадку полярність клітини регулюється шляхом стимулювання реорганізації цитоскелетона і зміни інтенсивності метаболізму кальцію.
Канонічний шлях – це послідовність реакцій, спрямованих на накопичення ß-catenin в клітині та її безпосередню доставку до нуклесу, де він буде діяти як транскрипційний фактор.
Шлях WNT починається з підключення WNT до рецептора для ліпопротеїну низької щільності (Low-density lipoprotein Relater протеїн-5, LRP-5), потім шляхом активування ефекту білка AXIN1 APC (негативний регулятор концентрації ß-catenin, мутації гена, що призводять до виникнення і розвитку кольорового раку), кінази GSK3, CK1 (кінази глікогедж).
Джерело: medach.pro/surgery/onkologiya-hirurgicheskie-distiplinyi/melanoma/























Сьогодні я отримав неправильний номер і випадково відправив мій батько повідомлення.
Мама син гіркий хліб